| Issue |
4open
Volume 5, 2022
|
|
|---|---|---|
| Article Number | 5 | |
| Number of page(s) | 27 | |
| Section | Life Sciences - Medicine | |
| DOI | https://doi.org/10.1051/fopen/2022002 | |
| Published online | 28 January 2022 | |
Review Article
Recent developments in mitochondrial medicine (part 2)
Aging, longevity and microbiota
1
Midwestern University, College of Pharmacy, Department of Pharmaceutical Sciences, 19555 N. 59th Avenue, Glendale, AZ 85308, USA
2
Université de Paris, INSERM U1016, Institut Cochin, CNRS UMR8104, Faculté de Médecine Cochin-Port Royal, 75014 Paris, France
3
Laboratory of Excellence GR-Ex, Paris 75015, France
* Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
29
October
2021
Accepted:
17
December
2021
Abstract
Called “bioblasts” in 1890, named “mitochondria” in 1898, baptized in 1957 as the “powerhouse of the cell” and christened in 1999 as the “motor of cell death”, mitochondria have been anointed in 2017 as “powerhouses of immunity”. In 1962, for the first time a causal link between mitochondria and human diseases was described, the genetic basis for which was revealed in 1988. The term “mitochondrial medicine” was coined in 1994. Research into mitochondria has been conducted ever since light microscopic studies during the end of the 19th century revealed their existence. To this day, new discoveries around this organelle and above all new insights into their fundamental role for human health and disease continue to surprise. Nowadays hardly any disease is known for which either the etiology or pathogenesis is not associated with malfunctioning mitochondria. In this second part of our review about recent developments in mitochondrial medicine we continue tracking and highlighting selected lines of mitochondrial research from their beginnings up to the present time. Mainly written for readers not familiar with this cell organelle, we hope both parts of our review will substantiate what we articulated over a decade ago, namely that the future of medicine will come through better understanding of the mitochondrion.
Key words: Mitochondrial Medicine / Aging / Microbiota / COVID-19 / Humanin / Reactive Oxygen Species
© V. Weissig & M. Edeas, Published by EDP Sciences, 2022
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
In this second part of our review about “Recent developments in mitochondrial medicine” (for Part 1, please see [1]) we continue to highlight selected lines of mitochondrial research from their beginnings up to the present time. Studies of this organelle can be traced back for over 170 years and to this day new revelations about basic mitochondrial functions and their implications for human health never cease to surprise.
Light microscopic studies on cells conducted over 150 years ago revealed the existence of small sub-cellular particles resembling in size and shape bacteria which Robert Altmann (1852-1900) named “bioblasts” in 1890 [2, 3]. Eight years later, Carl Bendas (1857–1932) coined the term “mitochondria” with “mitos” (Latin/Greek) meaning thread and “chondrion” (Latin/Greek) meaning grain, thereby properly describing their appearance on the microscopic slide. The following 50+ years of intense biological and biochemical research (see part 1 of our review [1]) culminated in the popularization of mitochondria as the “powerhouse of the cell” [4]. During the early 1960s the vital role of mitochondria for human health began to unfold, when Rolf Luft (1914–2007) described “a clinical case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial control…” [5]. It should be mentioned that it was also Rolf Luft, who in a retrospective paper in 1994 coined the term “Mitochondrial Medicine” [6]. Also in the early 1960s, Margit M.K. and Sylvan Nass discovered the existence of DNA inside mitochondria [7], which was sequenced in 1981 [8]. This in turn, during the late 1980s, opened up the way for unraveling the genetic basis for what has meanwhile become known as “Luft’s disease”: in 1988 Ian James Holt and co-workers described the link between mutations of mitochondrial DNA (mtDNA) and myopathies in a Nature paper [9] and Douglas Wallace and his team reported the association of mtDNA mutations with Leber’s Hereditary Optic Neuropathy in a Science paper [10]. The 1990s saw a boom in mitochondrial research. First, based on the pioneering work by Wallace and Holt and their co-workers, the number of diseases found to be associated with mutations and deletion of the mitochondrial genome literally exploded. Second, the pivotal role of mitochondria for “Programmed Cell Death” (apoptosis) has been revealed which prompted the pioneers of this line of research to christen mitochondria in 1999 as the “arsenal of the cell” or the “motor of cell death” [11]. Characterizing mitochondrial research during the 1990s as a booming field of biomedical science did not leave much room for finding a proper term to describe the last two decades, during which the number of publications on mitochondria has skyrocketed. In 2017, for example, mitochondria gained yet another moniker, when a review for Nature Immunology sanctified mitochondria as “powerhouses of immunity” [12] thus indicating that this organelle also seems to lie at the heart of immunological responses. In summary, nowadays there is little doubt that either the etiology or the pathogenesis of almost every disease appears to be linked to, or at least involves, malfunctioning or damaged mitochondria.
Mainly written for readers who are not familiar with the mitochondrial field of biomedical research we hope these two reviews will first succeed in conveying to the reader our fascination with this organelle and second substantiate what we have written over a decade ago, that “the future of medicine will come through mitochondria” [13].
Mitochondria and aging
Longevity, sometimes used as a synonym for “life expectancy”, was defined in 2006 by Benedictis and Franceschi as the capability to survive past the average age of death [14]. The human life expectancy and ageing outcomes have improved in the past century, largely as a consequence of improved social and environmental conditions and advanced medical care. Besides social and environmental factors also genes involved in different pathways of regulation, e.g. immune-inflammatory responses and regulation of oxidative stress, play a crucial role in “successful aging”. In particular, the impact of oxidative stress on healthy ageing and longevity is currently a hotly disputed topic in the field of age-related studies [15, 16] as we shall discuss below.
In 2013 mitochondrial dysfunction was acknowledged in a widely cited paper [17] as one out of nine “Hallmarks of Aging” (Fig. 1).
The idea of mitochondria being linked to biological aging, however, goes back to the 1950s. Denham Harman (1916–2014) proposed in 1956 his free radical theory of ageing based on the realization that reactive oxygen species (ROS) i.e. highly reactive free radicals are able to oxidatively damage biological macromolecules [18]. Such damage in turn would be the cause or at least significantly contribute to a general malfunctioning of cells, tissues and organs thereby bringing about aging. Haman wrote in his seminal paper that “aging and the degenerative diseases associated with it are attributed basically to the deleterious side attacks of free radicals on cell constituents and on the connected tissues” [18]. During the following years Haman transformed his free radical theory into the “Mitochondrial Free Radical Theory of Aging” (MFRTA) and the title of a paper he published in 1972 asked the question “A biologic clock: the mitochondria?” [19]. Haman’s conclusions were supported in the 1980s and 1990s among others by Miquel et al. [20], Sastre et al. [21] and Hagen et al. [22]. One major target for free radicals inside mitochondria is mtDNA. It should be mentioned here again (please also see Part 1 of our review) that mitochondria contain their own genome, which is a double-stranded circular DNA molecule of 16 569 bp. Originally described in 1963 by Margit M.K. Nass and Sylvan Nass as “intramitochondrial fibers with DNA characteristics” [23], it was in 1981 one of the first parts of the human genome to be sequenced [8]. All gene products of the mtDNA are essential for the synthesis and maintenance of the Oxidative Phosphorylation Electron Transfer Chain (OXPHOS-ETC). Consequently, any damage of mtDNA will result in the reduced production of adenosine triphosphate (ATP), i.e. the cellular energy currency. It has been proposed that the capacity for oxidative phosphorylation (i.e. for the generation of ATP) declines with age due to an increased load of defective mtDNA which also may play a role in the etiology of age-related neuromuscular and neurodegenerative diseases [24]. There is evidence that mtDNA damage seems to accumulate with age. For example, Cooper et al. described an age-related accumulation of the 5-kb common deletion of mtDNA in human skeletal muscle and in brain [25] and Hattori et al. found an age-dependent increase of a 7.4 kb deletion in mitochondrial DNA of the human heart [26]. In line with these findings, it could also be demonstrated that the overexpression of endogenous antioxidants, which are capable of neutralizing and removing free radicals, extends the life span of adult Drosophila melanogaster (commonly known as fruit fly), a widely used model organism in biological research [27, 28]. However, data demonstrating the opposite and thereby weakening the MFRTA appeared after 2000 For example, Huang et al. found that the overexpression of Copper–Zinc Superoxide Dismutase (CuZnSOD), an endogenous antioxidant, does not extend the life span in mice [29], which is not surprising considering that the end product of the SOD reaction, hydrogen peroxide, can only be detoxified by the seleno-enzyme Glutathione peroxidase [30]. Likewise, Van Remmen et al. showed in 2003 that although a life-long reduction of Manganese Superoxide Dismutase (MnSOD) activity, another endogenous antioxidant, leads to increased levels of oxidative DNA damage it does not appear to affect aging as one would expect [31]. Andziak et al. [32] compared in 2006 the antioxidant capacity and the level of oxidative damage in the naked mole-rat (Heterocephalus glaber) with similar-sized mice. Naked mole-rats are the longest living rodent species known, with an average life span of over 28 years their life is an order of magnitude longer than predicted based on their body size. Very surprisingly it was found that naked mole-rats have a significantly lower level of endogenous antioxidants and a much higher level of oxidative damage to lipids, DNA and proteins compared to “normal” weight-matched mice. The authors concluded that other mechanisms than attenuated oxidative stress must contribute to the exceptional longevity of this species [32] and two papers published in 2009 and 2012 were even asking in their title “is the oxidative stress theory of aging dead?” [33, 34]. Although an excess of free radicals (aka oxidative stress) can cause significant damage to cell components, it is nowadays accepted that the MFRTA alone is not sufficient to satisfactorily explain the complex process of aging. Multiple non-free radical mitochondrial mechanisms may also promote aging. For example, Sharma et al. reviewed recent studies that show how mitochondrial fission, fusion, and trafficking are altered with age. They discussed factors that change with age to directly or indirectly influence mitochondrial dynamics while examining roles for altered mitochondrial dynamics in healthy aging and underlying functional outputs that might affect longevity. Gathering data from past and emerging research, the studies highlighted in this review revealed that no single specific mitochondrial morphology state is pro‐ or anti‐aging. For example, fragmented mitochondria are directly linked to aging and short longevity in yeast, whereas the exact opposite was observed in the fruit fly in which fragmented mitochondria prevented ageing [35].
As if the connection between aging and mitochondria is not already complicated enough, yet another factor entered the scene, which even independently of its possible involvement in aging came as a big surprise. Since the sequencing of mtDNA in 1981 [8] we have known that mtDNA contains 37 genes, which encode 13 proteins, 22 tRNAs and 2rRNAs. But surprisingly, 20 years later, in 2001 Ikuo Nishimoto and his team from the Keio University of Medicine in Tokyo discovered a small peptide which was derived from a fragment of the 16S rRNA [36]. In other words, the mitochondrial gene for the 16S rRNA harbors the information for another gene product. This peptide named “humanin” is just 24 amino acids long and was found to have a wide range of neuroprotective properties [36–38]. A mitochondrial transcriptome analysis done 10 years later revealed the existence of several more small RNA molecules derived from mtDNA [39, 40]. As of last year, a total of eight so-called “mitochondrial-derived peptides” have been identified [41].
Back to humanin, in 2020 Yen et al. showed that humanin is a regulator of lifespan and health span. The authors have demonstrated that overexpression of humanin is sufficient to extend lifespan. Having established that humanin is sufficient to increase lifespan in worms (C. elegans), they examined if humanin treatment started in mid-life would it be able to increase lifespan in mice. Administration of humanin to female mice did not increase lifespan, but it was able to improve metabolic parameters (body size, weight, and fat) without changing food intake. They concluded that “humanin is not only correlated with health and lifespan but can significantly improve both parameters on its own” [42].
Finally, we refer the interested reader to a published review by Brandon Berry and Matt Kaeberlein [43] in which the authors make the case that mitochondrial dysfunction (i.e. dysregulation of mitochondrial metabolism) may contribute to all other hallmarks of aging and which in a reciprocal way may promote (is this a better expression?) malfunctioning of mitochondria. This intricate interaction between the nine hallmarks of aging, with perhaps mitochondria at the center, poses in our opinion a tremendous challenge to the ongoing endeavors aimed at slowing down, stopping or even reversing aging.
Mitochondrial quality control and age related diseases
At the Inaugural World Conference on Targeting Mitochondria in Berlin, Germany in 2010 Michael R. Duchen from University College of London emphasized that “…major neurodegenerative diseases of later life (Parkinson’s, Alzheimer’s…) are all associated with mitochondrial dysfunction..” and he further stated that “... in this context the notion of mitochondrial “Quality Control” has emerged as a key concept in mitochondrial biology” [44]. Mitochondrial quality control (MQC) refers to a complex system involving all factors and processes needed for maintaining a fully functioning and healthy population of mitochondria [45]. Qiu et al. define MQC as the “continuous biogenesis, fusion, fission and mitophagy (which) contribute(s) to the balance of mitochondria’s morphology, quantity and quality” [46]. Any disturbances within this system results in an accumulation of dysfunctional mitochondria over time and can contribute to the development of age-related diseases [47]. The mechanism for each of the three main processes responsible for sustaining a healthy mitochondrial population, i.e. biogenesis (formation of new mitochondria), dynamic (fission and fusion of mitochondria) and mitophagy (degradation and eliminating of damaged mitochondria) is currently under investigation. Any detailed discussion thereof is beyond the scope of this review. For an update about mitochondrial biogenesis please see [48], for mitochondrial dynamics [49] and for mitophagy [50]. Here we just would like to highlight a few aspects of interest.
Mitochondrial biogenesis
Any cell division requires cell organelles to double in size and divide to ensure the formation of two identical daughter cells. Mitochondria are no exception; they are formed by growth and division of existing organelles in coordination with the cell cycle [51]. However, mitochondria are also able to adapt to stress. This was observed for the first time during the 1960s, when John O. Holloszy (1933–2018) found that exercise training in rodents resulted in a significant increase of the mitochondrial mass in skeletal muscle [52, 53]. In his classic study from 1967 [52] Holloszy found a nearly doubled oxidative capacity, respiratory enzyme activity and mitochondrial biogenesis in rodent limb muscles after high-intensity running for 12 weeks, 5 days per week and 2 h per day. Subsequently, Holloszy and his group tried uncover the mechanisms for these adaptations, for example they were able to show that muscle contractions can increase an insulin-independent glucose transport into myocytes [54].
But it took about 40 years until the regulatory mechanisms behind the stress response Holloszy described during the 1960s began to become understand. In 1998, studying the regulation of adaptive thermogenesis in mice, Bruce Spiegelman’s group cloned a transcriptional coactivator from a brown fat cDNA library, which they named peroxisome proliferator-activated receptor-gamma coactivator 1 (PGC-1). It should perhaps be mentioned here that transcriptional co-activators do not bind directly to DNA as transcription factors do. But since regular transcription factors alone do not have the enzymatic activity to start translation of a particular gene, they need co-activators to do the job. Spiegelman’s PGC-1 was identified as a factor that interacts with a “regular” transcription factor called peroxisome proliferator-activated receptor gamma (PPAR-ϫ). In addition to PGC-1’s role in the response of mice to cold exposure, Spiegelman’s team also described the activation of key respiratory chain enzymes and most strikingly an increase of the cellular content of mitochondrial DNA following the ectopic expression of PGC-1 in white adipose cells [55].
Within less than a decade, Spiegelman’s PGC-1 was recognized and subsequently became known as the “master regulator of mitochondrial biogenesis” [56] PPAR-ϫ coactivator-1α (PGC-1α)”. More than 10 isoforms of PGC-1α displaying differential regulation and tissue distribution have been reported [57]. Currently, PGC-1α is considered the most important regulator of mitochondrial biogenesis, which can rapidly be induced under conditions of high energy consumption like cold, exercise and fasting [46, 58, 59].
Since the upregulation of mitochondrial biogenesis appears as a plausible strategy to overcome cellular energy deficit caused by malfunctioning mitochondria we would like to elaborate on this topic. Triggering the new formation of “additional” mitochondria (better increasing the mitochondrial mass) might even be a universal approach towards the therapy off all diseases associated with energy deficiency. Komen et al. stated that “boosting mitochondrial numbers would hypothetically increase the residual OXPHOS activity present in all patients” [60] suffering from mitochondrial diseases.
A few years after PGC-1α was cloned, a very close homolog, known as PGC-1β, was identified [61, 62]. Calling PGC-1α the master regulator of mitochondrial biogenesis might do some injustice to its close beta homolog, because both, PGC-1α and PGC-1β complement each other in ensuring mitochondrial maturation and function under varying physiological conditions. But in contrast to PGC-1α, the beta homolog does not appear to be involved in reacting to physiological stress like exercise or cold exposure. While knock-out mice with either interrupted PGC-1α or PGC-1β are viable, PGC-1α/β double knockout mice died due to cardiac failure within 24 h [63]. Strikingly, several laboratories confirmed that the overexpression of PGC-1α leads to an increase of mitochondrial mass [64–67]. Subsequently, PGC-1α has turned into an extremely valuable pharmacological target for increasing the mitochondrial mass in cells harboring dysfunctional mitochondria. One could argue that increasing the mitochondrial copy number in cells with malfunctioning mitochondria will only result in cells with more malfunctioning mitochondria. That might be true, but a larger number of malfunctional mitochondria together produce more ATP than cells with a smaller number of dysfunctional mitochondria. Komen and Thorburn wrote a few years ago in an excellent review which they appropriately named “Turn up the power – pharmacological activation of mitochondrial biogenesis…”: “…all patients with OXPHOS disorders have residual enzyme activity because a complete loss of activity is not compatible with life. Hence, up-regulation of the amount of less active mitochondria may increase ATP synthesis capacity above a certain threshold” [60] at which clinical symptoms caused by dysfunctional mitochondria could disappear. Since this is in our opinion such an important concept, we will add yet another level of detail to this review by briefly describing how the master regulator, PGC-1α, itself is being regulated or can be pharmacologically activated. Our explanation of PGC-1α control is mainly based on [58–60, 68–70] and Figure 2 has been adapted in a simplified form from [60].
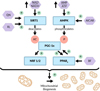 |
Figure 2 Schematic overview of factors controlling the activity of PGC-1α (adapted in a simplified form from [60].) QN, Quercetin; RL, Resveratrol; BF, Bezafibrate. |
PGC-1α already present in the cell (“constitutive” PGC-1α) can be activated or deactivated and additional PGC-1α can be synthesized following transcriptional activation. In the absence of physiological stress and during caloric abundance, constitutive PGC-1α exists in an inactivated acetylated state. Exercise as well as caloric restriction use up existing ATP which leads to an increased AMP/ATP ratio. To replenish ATP, reduced Nicotinamide Adenine Dinucleotide (NADH) is oxidized by OXPHOS which leads to an increased NAD+/NADH ratio. Both of these metabolite ratios control the activity of constitutive PGC-1α. An increased ratio of NAD+/NADH activates Sirtuin 1 (SIRT 1), an enzyme which deacetylates and thereby switches on PGC-1α. Sirtuins are a large group of enzymes/proteins which have been discovered just during the last two decades (please see below). Interestingly from a nutritional point of view, SIRT 1 can also be activated by quercetin, a polyphenol found in fruits and vegetables as well as by resveratrol, a stilbenoid mainly found in a variety of berries and the skin of grapes. On the other hand, an increased AMP/ATP ratio activates AMP-Kinase (AMPK) which phosphorylates and thereby also activates PGC-1α. Intriguingly, a long-known drug, 5-aminoimdazole-4-carboxamide ribonucleoside (AICAR), used since the late 1980’s to protect against cardiac ischemic injury [71], also activates AMPK. Noteworthy, AICAR has become notorious when it was used during the 2009 Tour de-France as a performance-enhancing drug and has subsequently been banned by the World Anti-Doping Agency [72, 73].
So, what is the role of activated PGC-1α? As an activated transcriptional co-activator it binds to and activates a variety of “regular” transcription factors which in turn launch the transcription and translation of a large number of genes needed for the production of additional mitochondria. To simplify, only two groups of those transcription factors will be introduced, Nuclear Respiratory Factors 1 and 2 (NRF1 and NRF2) and PPARs (Fig. 2). NRF 1 and 2 are transcription factors which control the gene expression of a large number of diverse proteins and enzymes needed for metabolism, cellular growth and stress responses. During the past decade, NRF2 has been recognized for its essential role in maintaining mitochondrial integrity and function. Moreover, experimental data suggest that NRF2 activity may have a direct effect on mitochondrial biogenesis [74–76]. Therefore, not surprisingly, NRF2 is the pharmacological target of RTA 408, a synthetic triterpenoid, which is currently tested in clinical trials for the treatment of mitochondrial myopathy and Friedreich’s ataxia [77]. The second group of transcription factors activated by PGC-1α involves at least three different PPARs which activate the transcription of a large number of genes encoding a multitude of proteins essential for cellular function, noteworthy including PGC-1α itself. Bezafibrate, is in clinical use since the 1970s as a lipid-lowering drug for the treatment of hyperlipidemia has surprisingly been found to be able to activate PPAR-regulated gene expression including the expression of PGC-1. Consequently, based on its ability to induce mitochondrial biogenesis, Bezafibrate has recently been discussed as a drug candidate for improving osteogenesis in patients with Leigh’s syndrome [78].
Mitochondrial dynamics
The discovery of “mitochondrial dynamics” dates back to 1914 when Margaret Adaline Reed Lewis (1881–1970) and her husband, Warren Harmon Lewis (1870–1964) observed frequent changes in the appearance of mitochondria in living cells. In their paper from 1914 they wrote: “The mitochondria are almost never at rest… the changes in shape are truly remarkable not only in the great variety of forms, but also in the rapidity with which they change from one form to another” [79]. In a more detailed follow-up paper from 1915 Margaret Lewis published a time-lapse study covering a period of 25 minutes during which she tried to capture how mitochondria constantly change their shape by drawing them with her own hand. In one of the figure’s legend she wrote “The changes were so rapid that it was not always possible to draw each mitochondrium” [80]. Amazingly, these early observations seem to have been largely forgotten or ignored until the beginning of the 2000s when the mechanism of fission and fusion as well as the importance of mitochondrial dynamic for human diseases began to be discovered [81–87]. Two recent very comprehensive reviews covering mitochondrial dynamics [49, 88] do not cite any work done prior to 1998 (except in [88], where one reference from 1986 dedicated to the mitochondrial genome is cited). Therefore, to us it does not appear as exaggerated to claim that there has been an explosion of knowledge during the last two decades about the mechanism of mitochondrial fission and fusion and at the same time about the involvement of mitochondrial dynamics in etiology and pathogenesis of human diseases. What we know today, has recently been summarized by the pioneer of studying mitochondrial dynamics in mammalian cells, David C. Chan (California Institute of Technology, Pasadena, CA) in [49]: first, the dynamic behavior of mitochondria is essential for their function; second, the key mediators of fission and fusion have been identified as GTP using enzymes that perform mechanical work on lipid bilayers; third, several neurodegenerative diseases (e.g. Parkinson’s, Alzheimer’s) are associated with mutations in genes encoding for those enzymes needed for fusion and fission; Fourth, even some common diseases like cancers are associated with mitochondrial dynamic, but the causal link still has to be determined [49].
Mitophagy
An essential component of MQC, i.e. of maintaining a healthy population of mitochondria is the degradation and removal of damaged mitochondria. The presence of degraded mitochondria in cells was reported in 1915 by Margaret Lewis in the same paper in which she described the constant change of shape of these organelles [80]. She wrote over a hundred years ago: “Some cells are found which contain many normal mitochondria, some partly degenerate, and others entirely degenerate… It is apparent that some change has taken place which has completely changed not only the morphology but also the composition of the mitochondria” [80].
They key to unlocking the secret surrounding the demise of mitochondria as observed by Margaret Lewis was provided 40 years later by Christian de Duve (1917–2013; Nobel Prize in Physiology or Medicine in 1974) who serendipitously discovered cytosolic particles which he named in 1955 “lysosomes”, Greek for “digestive bodies” [89]. With financial support from the Lilly Research Laboratories Christian De Duve’s major goal was to investigate the action of insulin on the liver. In the course of these studies, he focused on the characterization of glucose-6-phosphatase, an enzyme now known to be responsible for the releases of glucose from hepatocytes following gluconeogenesis. To distinguish this specific phosphatase he had to separate it from a non-specific acid phosphatase which caused experimental problems since this enzyme turned out to be trapped in “sac-like particles surrounded by a membrane” [90]. Follow-up experiments revealed more insight into these sac-like particles [91] nowadays known as lysosomes and in retrospective, De Duve wrote in 2005 “… we were hooked… insulin and carbohydrate metabolism were abandoned” [92]. Though de Duve’s serendipitous discovery of lysosomes is not directly linked to the topic of our review, we would like to quote from the same paper his reflection about the value of serendipity we deem still to be of high importance: “My final point concerns the value of serendipity and, especially, of its necessary correlate, the freedom to follow a new trail opened by chance, irrespective of previous commitments. It is noteworthy that in the paper in which lysosomes are first mentioned, the Lilly Research Laboratories are acknowledged for their financial support, despite the fact that their major interest, insulin, is conspicuously absent from the paper. One could only wish that funding agencies might show the same liberal attitude today” [92].
Going back to Margaret Lewis’ observed demise of mitochondria, in 1962 Thomas P. Ashford and Keith R. Porter discovered mitochondrial fragments in liver lysosomes using electron microscopy. They wrote “One may reasonably conclude that the mitochondria are in various stages of breakdown or hydrolysis” and they called this process “autolysis” [93]. The term “autolysis” was replaced five years later by Christian de Duve who proposed “autophagy” derived from the Greek meaning of “eating of self” [94, 95]. For the following 40 years the process of cytoplasm being sequestered into autophagosomes for subsequent delivery to and fusion with lysosomes for degrading macromolecules and cell organelles was thought to be a random process. This view changed when Nadine Camougrand and her colleagues from the University of Bordeaux were able to demonstrate in 2004 that in yeast a specific membrane protein in the mitochondrial outer membrane was essential for mitochondrial autophagy. Mutation of this protein suppressed autophagy of mitochondria without any impact on autophagic degradation of other cytosolic components [96, 97].
Subsequently, one year later, John J. Lehmasters from the University of North Carolina at Chapel Hill proposed in 2005 the new term “mitophagy” “… to emphasize the non-random nature of the process” [98].
Considering the vital role of mitophagy for maintaining a healthy population of fully functioning mitochondria (see MQC above) it is not surprising that mitophagy has emerged as an important potential new therapeutic target. During the last decade, a number of low-molecular compounds as well as lifestyle interventions have been found to modulate mitophagy and subsequently to positively affect health and lifespan. This new field of biomedical research (modulating mitophagy) has recently been comprehensively reviewed by Daniela Bakula and Morten Scheibye-Knudsen from the University of Copenhagen who coined the new term “mitophaging” in [50]). The authors wrote in their concluding remarks: “… given the central role of mitophaging in multiple age-related pathologies it appears highly likely that these new promising approaches may present possible interventions in age-related diseases. The future is bright!” [50]. Only a few of these “new promising approaches” will be discussed. First, lifestyle interventions which have a positive impact on mitophagy (or on “mitophaging”) are caloric restriction and exercise [50]. Second, one of the best studied small molecules in term of lifespan extension and disease development in animal models are rapamycin and its analogs, called rapalogs [99]. Rapamycin is a naturally occurring macrolide, i.e. a large cyclic lactone ring with conjugated sugars, which was isolated in 1972 from a specific bacterium found on Easter Island [100]. It was approved by the FDA (USA) in 1999 as an immunosuppressive and is being traded under the name Rapamune (Pfizer). The study of this potent immunosuppressive and anti-proliferative compound actually led to the discovery of whole new signaling pathway triggered by mTOR, which stands for mechanistic (mammalian) target of rapamycin [101, 102]. This “mechanistic target of rapamycin (mTOR)” represents a large group of protein kinases, which are enzymes that activate or deactivate proteins and other enzymes involved in fundamental cellular processes like growth, proliferation, differentiation, motility and others [103, 104].
Other metabolites and enzymes affecting mitophagy are NAD+, SIRT1 and AMPK [50], which were discussed above for their role in mitochondrial biogenesis. As reviewed in [50], numerous mitophagy modulators have been identified, many of them are naturally occurring compounds. The big challenge for pharmacologically augmenting mitophagy however is associated with the specific targeting of damaged mitochondria. Whether the future of mitophagy modulators for lifespan extension and healthy aging is bright will depend on clinical trials yet to be designed and conducted.
Mitochondrial unfolded protein response (UPRmt)
Transmembrane and secretory proteins undergo posttranslational protein modification and subsequently fold into their native conformation inside the endoplasmic reticulum (ER). The ER is also the site of biosynthesis for steroids including cholesterol and many lipids which requires the presence of numerous enzymes. Subsequently, the total ER protein concentration can reach up to 100 mg/mL [105] (for comparison, the protein concentration in blood serum is 60–80 mg/mL). To maintain full functionality of all proteins present inside the ER at any given time and to prevent aggregation and accumulation of misfolded proteins a system of chaperone molecules has evolved which uses metabolic energy to ensure proper protein folding. An increase in the level of misfolded proteins triggers signaling pathways which leads to upregulation of chaperon expression. The first investigations into these chaperon proteins and into this pathway were launched at the beginning of the mid-1970s [106]. By the beginning of the 1990s this signaling pathway has been designated as “unfolded protein response (UPR)” [106, 107] and by the beginning of the 2000s malfunctioning UPRs have been linked to a variety of human diseases [105].
Considering the very high protein concentration inside mitochondria (in hepatocyte mitochondria up to 500 mg/mL [108]), Nick Hoogenraad and colleagues started in the mid-1990s to investigate whether mitochondria possess a similar signaling pathway which can be triggered by an increasing load of misfolded mitochondrial proteins [109]. Subsequently, between 1996 and 2012, this group has published a series of papers demonstrating that mitochondria possess their own unfolded protein response, termed UPRmt [110–114]. Though the UPRER and the UPRmt share common elements, controlling factors are entirely different [109]. Current efforts focus on the further elucidation of the UPRmt mechanism and on its exploration as a potential therapeutic target [109, 115, 116].
What is the nuclear factor kappa B (NF-kB) doing in mitochondria?
During the mid1980’s Ranjan Sen working in the laboratory of Nobel laureate David Baltimore identified a protein that binds to a specific and conserved DNA sequence in nuclei of activated B lymphocytes, which he named “Nuclear Factor binding near the k light chain gene in B cells”, abbreviated as NF-kB [117, 118]. We know now that NF-kB involves a whole family of related protein complexes which exist either as hetero- or homodimers drawing from a pool of 5 different monomers resulting in the formation of up to 15 different NF-kB complexes [118–120]. The role of NF-kB in the proper functioning of cells cannot be underestimated; by either activating or repressing “hundreds of target genes” [118] it controls DNA transcription, cytokine production, cell survival and other fundamental cellular events. In particular NF-kB is involved in regulating the immune response to infection. As of today, the diverse family of NF-kB transcription factors “has been extensively characterized in organisms ranging from flies to humans” [121] and their recent discovery in basal phyla including sea anemones, corals, hydras, jellyfish and sponges has triggered further investigations into the evolutionary roots of this group of transcription factors which is expected to lead to a better understanding of the origins of regulated innate immunity [121]. Also interesting, inhibitors of NF-kB signal transduction pathways are considered as promising anti-inflammatory therapeutics [122].
So, what is the connection between NF-kB and mitochondria? It is to the credit of Benedict C. Albensi (since June 2021 at Nova Southeast University in Florida) who drew the attention of the mitochondrial community most recently to this “master regulator of evolutionarily conserved biochemical cascades” [123] by a paper with the thought-provoking title “What is NF-kB doing in and to the mitochondrion?” [124]. Though first reports about NF-kB subunits having been found in purified mitochondria were published in 2001 [125, 126] and three confirming papers appeared shortly thereafter [127–129], “not much follow-up work has been done to date” [124] which is “surprising” [124] considering the fundamental role and the evolutionary history of this transcription factor family and of mitochondria. Albensi then discusses, on occasion in a speculative and perhaps even provocative way (in his own judgement), possible connections between NF-kB and mitochondrial dynamic, mitochondria-based apoptosis, mitochondrial respiration, mitochondrial metabolism and even mitochondrial gene expression. At the end of his review paper, Albensi is re-asking the question “What is NF-kB doing in and to the mitochondrion?” and gives the immediate and short reply “a lot!” [124]. It does not appear as unreasonable to assume that during the next decade NF-kB will get into the limelight of mitochondrial research. Currently (please see below) NF-kB is already being explored as a therapeutic target for the treatment of mitochondrial diseases.
The emerging role of NRF2 in mitochondrial bioenergetics
At the 6th World Congress on Targeting Mitochondria in October 2015 in Berlin, Germany, Albena Dinkova-Kostova from the University of Dundee gave a presentation about the increasingly recognized importance of the KEAP/NRF2 pathway for mitochondrial physiology and bioenergetics. NRF2 was firstly discovered and described in 1994 as a basic leucine zipper transcription factor with unclear biological function [130]. Two years later, studies of NRF2 knockout mice were unable to detect any abnormal phenotype of these mice but revealed that they were susceptible to stress [131]. In 1997, it was revealed that NRF2 knockout mice were unable to induce the expression of two antioxidant enzymes [132]. Today we know that NRF2 is a transcription factor for over 500 genes, most of which have cytoprotective functions. NRF2 regulates a large variety of genes that are crucial for controlling cellular resistance to intrinsic and extrinsic stressors [133–135]. In particular, it has been demonstrated that NRF2 stimulates mitochondrial biogenesis by promoting purine nucleotide biosynthesis and by controlling the levels of nuclear respiratory factor 1 and peroxisome proliferator-activated receptor ϫ coactivator 1α [75]. In summary, NRF2 has emerged as an important factor for maintaining the structural integrity of mitochondria [75] and its activation appears to generally improve mitochondrial function [76] which makes it a prime therapeutic target for the treatment of mitochondrial diseases (see below).
NRF2 and NF-kB as therapeutic targets for the treatment of mitochondrial diseases
Both, the NRF2 and the NF-kB pathways are increasingly being discussed as potential therapeutic targets for the treatment of a variety of clinical conditions [135–145]. With respect to the drug development for the therapy of mitochondrial diseases (extensively reviewed in [77]) one potential new drug (New Chemical Entity, NCE) will be highlighted. RTA 408 (Omaveloxolone) is a synthetic oleanane triterpenoid which has been shown to activate NRF2 and to suppress NF-kB [146]. This NCE has undergone one Phase II clinical trials (completed in 2017) for the treatment of mitochondrial myopathy and is currently being tested in an ongoing Phase II clinical trial for the treatment of Friedreich’s ataxia [77]. In vitro studies conducted with cells isolated from patients have shown (based on mitochondrial membrane potential measurements) that RTA 408-mediated activation of NRF2 increases mitochondrial functions [146]. It appears that RTA 408 might be able to improve the generation of ATP and at the same time may suppress uncontrolled inflammatory responses [77]. Preliminarily released data from a cohort of 69 Friedreich’s ataxia patients show that NCE is well tolerated and at an optimal dose of 160 mg/day appears to improve neurological functions [146].
Mitochondrial sirtuins
Sirtuins are a group of enzymes which act as essential signaling proteins in large number of fundamental cellular processes. The name “sirtuin” has been derived from its “founding member” [147] Sir2, which stands for “Silent Information Regulator” [148]. The entire field of sirtuins exploded during the last 20–25 years, not least due a study which showed that the overexpression of one particular sirtuin significantly increases the lifespan of yeast [149], a finding which was soon thereafter confirmed in worms [150] and in flies [151]. But the effect of sirtuins on lifespan extension has been under dispute [147]. For example, sirtuin-activating compounds were found to slow-down aging in mice and non-human primates [152], but corresponding clinical trials were inconclusive [153]. Any discussion of inconsistencies around the effects of sirtuins on lifespan is however beyond the scope of our review.
Most sirtuins act as deacylases, which are enzymes which remove either acetyl, malonyl or succinyl residues from the amino acid lysine in target proteins and thereby regulate protein function and activities. In contrast to “classical” deacylases however, sirtuins couple the deacylation process to NAD+ hydrolysis thereby generating free nicotinamide. A few sirtuins function as ADP-ribosyltransferases, i.e. they conjugate the ADP-ribose moiety to lysines in proteins.
Since the activity of all sirtuins depends on the availability of NAD+, all post-translational sirtuin – based protein modifications are directly coupled to the energy state of the cell. It has been found that several factors (not discussed here) may play a role in the observed age-related decline of NAD+ levels in humans [154, 155] and it has been hypothesized that “NAD(+) depletion may play a major role in the aging process, by limiting energy production, DNA repair and genomic signaling” [154].
Seven distinct sirtuins have been identified in mammals, named SIRT1 to SIRT7; three of them, SIRT 3, 4 and 5 are localized inside mitochondria. The functions and detailed mechanisms of actions of all three mitochondrial sirtuins have been recently and comprehensively reviewed by Chih-Hao Wang and Yau-Huei Wei, respectively [156]. In brief, SIRT3, SIRT4 and SIRT5 regulate in a highly coordinated fashion, via activation or deactivation of numerous key enzymes, cellular metabolism (fatty acid oxidation, TCA cycle, urea cycle), redox homeostasis (antioxidant activities) and the OXPHOS pathway (ATP production). Defects related to all three mitochondrial sirtuins have also been postulated to contribute to the pathogenesis of insulin resistance and type 2 diabetes (T2D) [156].
In summary, mitochondrial sirtuins play an important role and their dependence on the presence of cellular NAD+ which declines with age, dietary supplementation with NAD+ precursors could also be considered.
The changing dogma about mitochondrial Reactive Oxygen Species
Equally famous for being the powerhouse of the cell, mitochondria have gained and persistently retain the reputation of being the major source of highly toxic Reactive Oxygen Species (ROS). The notion that electrons leak from the mitochondrial electron transfer chain to reduce oxygen to superoxide radicals which is converted by superoxide dismutase to hydrogen peroxide that in turn and in the presence of free Fe2+ forms highly reactive hydroxyl radicals has become common textbook knowledge. Even an Editorial from 2015 still considers ROS as nothing but being “detrimental by-products” of cellular respiration [157].
The presence of strong intracellular oxidants of course can cause non-specific oxidative damage to nucleic acids, lipids and proteins. The identification of such “oxidative stress” as a major factor for the etiology and pathogenesis of a wide spectrum of human diseases including cancer, Alzheimer’s, Parkinson’s diabetes and aging has led to the general embracement of all types of natural and synthetic antioxidants for prophylaxis and therapy of human disease states which in turn gave rise over the last three decades to a large branch of the supplement industry. Remarkably, despite the straightforward sounding concept of fighting toxic oxidants by administering antioxidants the literature is filled with reports about failed antioxidant therapy studies (reviewed in [158]).
So, how did mitochondria gain their reputation as the major source of toxic ROS and what has changed during the last 20 years?
Intact mitochondria have been established as a novel source of intracellular hydrogen peroxide at the beginning of the 1970s. At that time “the possibility of a respiratory chain linked hydrogen peroxide production under physiological conditions” has been considered for the first time [159, 160]. A series of follow-up in vitro studies were summarized in 1979 in an influential review paper about hydroperoxide metabolism in mammalian cells [161] in which the author concluded from the data available that approximately 2% of the total inspired oxygen is converted to hydrogen peroxide. This conclusion remained unchallenged for over 20 years and the concept that ROS are highly toxic agents which cause the random oxidation of macromolecules became a dogma.
It was Anthony William Linnane (1930–2017) and colleagues who started in the early 2000s to question this supposition by compiling substantial evidence of the vital requirement for the “continuous regulated formation and functional utilization of superoxide anion and hydrogen peroxide, as well as the free radical gas nitric oxide and peroxynitrite” [162–165]. According to Linnane [164], St-Pierre and his group reported in 2002 [166] that the earlier studies significantly overestimated the amount of superoxide anions and hydrogen peroxide by about two orders of magnitude. Instead of 10 nM H2O2/minute/mg mitochondrial protein only about 0.1 nM H2O2/minute/mg mitochondrial protein is formed under physiological conditions [164]. The papers by Linnane and by St-Pierre and additional papers by Nohl [167, 168] show that inhibited respiring mitochondria produce only very low levels of ROS and that under physiological conditions hydrogen peroxide is not as toxic as its reputation, except at elevated concentrations reaching the mM range.
In summary, the developments made in redox biology since around 2000 have revealed the vital function of ROS and in particular hydrogen peroxide as messenger molecules essential for the regulation of the entire metabolome. Cellular signaling by ROS and Reactive Nitrogen Species RNS is now generally acknowledged as a major and essential physiological process [169, 170]. Whether ROS are toxic or not largely depends on their intracellular concentrations. It has been suggested to distinguish between three levels of ROS/RNS (Fig. 3), each one playing different biological roles: i) a “physiologic level” at which ROS/RNS are essential components of redox-sensitive signaling pathways; ii) “elevated levels” arising from cellular stress preparing the cell for survival, and iii) “pathologic levels” at which ROS or RNS lead to massive oxidative damage of cellular components resulting in cell death [171].
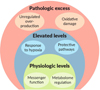 |
Figure 3 Scheme illustrating functions and effects of three different levels of the intracellular concentrations of reactive oxygen/nitrogen species (adapted from [158]) |
In conclusion, for any successful antioxidant therapy, delivery technologies have to be developed which are aimed at neutralizing only the excess of ROS/RNS, but this is beyond the scope of this review.
Targeting mitochondria with low molecular weight drugs
A 1969 paper from by Vladimir Skulachev’s group in Moscow (Russia) described the membrane potential-dependent accumulation of methyl-triphenylphosphonium (MTPP) cations inside the matrix of mammalian mitochondria [172]. MTPP is an amphiphilic molecule with a delocalized positive charge, also simply called a delocalized cation (DLC). During the 1980s similar amphiphilic molecules with a delocalized positive charge (e.g. rhodamine 123, dequalinium) were found to localize inside respiring mitochondria [173–176] which lead to the dogma that amphiphilicity paired with a delocalized positive charge center is the ultimate prerequisite for inherent mitochondriotropism of low-molecular weight compounds. Sufficient lipophilicity in combination with the delocalization of the charge center to reduce the free energy change when moving from an aqueous to a hydrophobic environment were believed to be pre a requisite for mitochondrial accumulation of DLCs in response to the mitochondrial membrane potential [176]. However, the nature of “mitochondriotropism” of low-molecular weight compounds appears to be more complex. In 2007, a non-biased sample of over 100 mitochondriotropics (i.e. compounds reported to accumulate at or inside mitochondria) was generated from the literature and examined using physicochemical classification as well as the combination of quantitative structure-activity relationship (QSAR) models with a Fick–Nernst–Planck physicochemical model. Thus although DLCs have been regarded as the commonest structural type, only a third were such. Much the same proportion were acids, potentially or actually anions and many mitochondriotropics were electrically neutral compounds [177]. In conclusion, it can be said that all DLC are mitochondriotropics, but not all mitochondriotropics are DLCs.
The inherent mitochondriotropism of Skulachev’s MTPP was explored during the 1990s by Mike Murphy and his group to deliver a large variety of small molecules via conjugation to triphenylphosphonium cations into mammalian mitochondria [178–184], among them the active antioxidant moiety of vitamin E. Murphy and co-workers could show that the mitochondrially targeted vitamin E derivative accumulates several hundred-fold inside the mitochondrial matrix, prevents lipid peroxidation and thereby protects mitochondria from oxidative damage [183, 185]. Follow-up work eventually led to the development of MitoQ, which underwent four clinical trials over the past five years for the treatment of mitochondrial diseases as well as five more trials listed for treatment of other conditions [77]. A well-proven safety profile and first indications of therapeutic efficacy seem to make MitoQ a promising new drug for the therapy of a variety of diseases associated with an increased oxidative stress. Structurally closely related to MitoQ is SKQ1, developed by Vladimir Skulachev himself (S and K are the two first letters of the inventor’s last name). SKQ1 has been tested in two clinical trials in the US for the potential treatment of Dry Eye Disease (DED). It has been hypothesized that mitochondria-induced oxidative damage in the lacrimal gland is associated with the development of DED. In Russia this drug has already received a market approval.
Mitochondria-targeted peptides
A serendipitous discovery at the beginning of the 2000s laid the ground for the development of a potentially potent drug for the therapy of mitochondrial disorders. While developing peptides displaying high affinity and selectivity for the μ-opioid receptor it was discovered rather surprisingly [186] that a few of those synthetic peptides, among them the tetrapeptide MTP-131, accumulated at the inner mitochondria membrane and even prevented mitochondrial swelling and oxidative cell death during reperfusion injury [187]. Further investigations revealed that these mitochondriotropic peptides selectively bind to cardiolipin (CL), a phospholipid specific for the inner mitochondrial membrane. As a result of that binding the interaction between cytochrome C and CL appears to be modulated which in turn seems to accelerate electron flow through the electron transfer chain (ETC) [186, 188]. It is hypothesized that the support of the electron transport through the ETC contributes to an increased ATP output [189, 190]. During the last 5 years, MTP-131 (“Elamipretide”) was tested in eight clinical trials for a variety of clinical conditions [77]. In general, it appears that this new experimental drug is safe and seems to exert a supporting effect on mitochondrial function in disease states associated with malfunctioning mitochondria. Its discovery, again, was purely accidental.
Mitochondrial ion channels
Mitochondrial ATP production through oxidative phosphorylation requires a mitochondrial inner membrane essentially impermeable to ions in order to minimize energy dissipation. Although by the middle of the 2000s plenty of evidence for the existence of mitochondrial ion channels has been accumulated, mitochondrial inner membrane channels have been considered still as a “nascent subject of investigations” [191]. Since a comprehensive discussion of this particular field is beyond the limits of this review, only a few selected examples illustrating the history and significance of research about mitochondrial ion channels will be reviewed below. For an extensive review of mitochondrial ion channels the reader is referred to [191, 192].
Voltage-dependent anion channel
As one of the earlier identified channels the Voltage-Dependent Anion Channel (VDAC) was purified from paramecium mitochondria and reconstituted in liposomes in 1976 [193]. It is a highly conserved protein with homology to bacterial porins and forms an outer membrane pore with a diameter of up to 3 nm. It allows the passage of ATP, ADP, phosphate, as well as Ca2+, K+ and Na+. The exact structure of VDAC, its interactions with a variety of other mitochondrial proteins and above all its role in mitochondrial pathologies including apoptosis is still under intense investigation. For example, a paper published in 2005, i.e. 30 years after the discovery of VDAC, carried the title: “On the role of VDAC in apoptosis: Fact and Fiction” [194]. A most recent review entitled “Renaissance of VDAC: New insights…” [195] stated that it “has become impossible to review all the existing literature on Voltage-Dependent Anion selective Channel (VDAC) in a single article”.
Uncoupling proteins
To better understand the function of uncoupling proteins (UCPs), a brief refresher of the mechanism of mitochondrial ATP production might be helpful. During glycolysis, beta-oxidation and the citrate cycle, hydrogen in the form of NADH2 and FADH2 is extracted from glucose and fatty acids. The subsequent oxidation of hydrogen atoms to form water, i.e. the transfer of electrons to molecular oxygen takes place at the respiratory complex at the mitochondrial inner membrane (see also Part 1 under “From Atmungsferment to Respirasomes”). When done in vitro, the oxidation of hydrogen to water is an explosive process (“Knallgas” reaction), but during the step-by-step transfer of electrons along the complexes of the ETC the energy is gradually released and used to build up a proton gradient across the highly impermeable inner mitochondrial membrane (IMM). Finally, protons move back into the mitochondrial matrix through the ATP Synthase (complex V), which in turn produces ATP via phosphorylation of ADP. During the entire pathway of oxidative phosphorylation, chemical energy (glucose, NADH2, fatty acids, FADH2) is converted first into redox energy (electron flow), then electroosmotic energy (proton gradient) and finally again into chemical energy (ATP). In this process the electron flow through the ETC is “coupled” with the production of ATP. Any opening of the inner mitochondrial membrane would allow protons to move back into the mitochondrial matrix bypassing complex V. Instead of being stored as chemical energy in form of ATP the electroosmotic energy would then be released as heat energy. Under such conditions, the electron flow is considered being “uncoupled” from the ATP production and that is exactly what a mitochondrial uncoupling protein does: “(It) provides a carefully regulated proton re-entry pathway across the mitochondrial inner membrane operating in parallel to the ATP synthase” [196]. As result, the rate of respiration increases in order to produce heat energy in addition to chemical energy in form of ATP.
The observation that brown adipose tissue is potentially able to produce more heat than other tissues has been made by several investigators during the 1960s [197]. The mechanism behind this phenomenon began to unfoldin 1978 when David G. Nicholls (today Professor Emeritus of Mitochondrial Physiology at the Buck Institute in Novato, CA) identified a 32 kDa protein in the IMM which is able to facilitate the transfer of protons back from the intermembrane space into the mitochondrial matrix [198]. This protein was originally named uncoupling protein, it also became known as “Thermogenin”, eventually was called Uncoupling Protein 1 (UCP1) and it was cloned for the first time in 1988 [199, 200]. Although by 2012 four more isoforms of UCP1 had been communicated in the literature (UCP2-5) [201–204], their discoveries were apparently not free of controversies. In a most recent review from 2021, David G. Nicholls wrote: “In the 40 years since UCP1 was first described, an extensive, and frequently contradictory, literature has accumulated… finally, the troubled history of the putative novel uncoupling proteins, UCP2 and UCP3, will be evaluated” [196]. Any detailed discussion of this dispute is however beyond the scope of our review.
Mitochondrial Ca2+ channels
Mitochondrial Ca2+ uptake has described for the first time in the early 1960s [205]. Since then the importance of mitochondrial Ca2+ homeostasis for cellular physiology and pathophysiology has been widely recognized (reviewed in [206]). Yet the molecular structure and identity of mitochondrial Ca2+ channels and/or transporters has remained elusive until around 2010 when two papers described a comparative genomic approach which led to the identification of two channels, called “Mitochondrial Calcium Uptake 1” (MICU1) [207] and “Mitochondrial Calcium Uniporter” (MCU) [208]. Fabiana Perocchi presented both of her papers at the 3rd World Congress on Targeting Mitochondria in Berlin, Germany and her presentation had the subtitle “50-year search for Calcium channels ends”. During the same time, De Stefani and colleagues described in another paper in 2011 the identification of the MCU as a 40-kDa protein located in the inner mitochondrial membrane (IMM)[209]. Studies of the exact mechanisms of how mitochondria regulate cellular Ca2+ homeostasis are ongoing. For example, most recently, in 2020, detailed structures of the human MCU in the presence and absence of Ca2+ have been published [210], a discussion thereof is however beyond the scope of our review. Any impairment of the proper function of the MCU can have catastrophic consequences. Noemi Esteras and Andrey Abramov from the UCL Queen Square Institute of Neurology in London, UK, comprehensively reviewed available data which show a strong link between malfunctioning MCUs and neurological disease such as Alzheimer’s Disease (AD) [211]. Briefly, two histopathological hallmarks of AD are the appearances of aggregates of misfolded proteins (beta-amyloid and tau) inside and outside neurons. Recent studies point at a direct role of these protein aggregates in disrupting the mitochondrial influx and efflux of calcium which in turn triggers cell death by mitochondria-mediated apoptosis. The recognition of such specific mechanism may lead to new mitochondria-targeted therapies for AD and similar neurological disorders [211].
K+ channels
The first mitochondrial inner membrane K+ channel (mitoKATP) was identified in 1991 using the direct mitoplast patch-clamp method [212]. Mitoplasts are mitochondria stripped off their outer membrane. The same technology revealed in 1999 the existence of a second channel (mitoKCa) [213]. As the nomenclature indicates, the first one is ATP sensitive, while the second one is controlled by calcium. Both potassium channels are thought to play a role (among others) for cellular tolerance to ischemia-reperfusion injury. Already back in 1985 it was demonstrated that agents able to open K+ channels in the plasma membrane of myocardial cells can protect hearts against ischemia-reperfusion injury [214]. Currently, mitochondrial K+ channels are being investigated as drug targets for cardioprotection [215–217].
Bidirectional cross-talk between mitochondria and microbiota: physiology and implication in many diseases
The human body is inhabited, from birth on, by millions of small microorganisms, which all together form the human microbiota. This microbiota, itself made up by millions of bacteria, fungi, and viruses, are shown to be vital for the well-being of the human body. The biggest populations of microorganisms inhabit the gut, while other popular places for microbiota are the skin, the genitals, and the respiratory tract. The healthy microbiota is of a vital importance for nutrition, immunity, and can also affect the metabolic and the neurobehavioral state of humans.
More in depth, the gut microbiota is an important plus for the human body in terms of fermenting the non-digestible dietary fibers, endogenous mucous, and others. The most important byproducts of the colonic microbial fermentation are urolithin A, and short chain fatty acids (SCFAs), primarily acetate, propionate, and butyrate [218]. Due to these metabolites, among others, the microbiota interacts with host cells in particular, more precisely by interlacing with the mitochondrial activities. In fact, this microbiota–mitochondria interplay is not surprising due to the features they share in common. In 1967, as we pointed out in Part 1 of our review [219], Lynn Margulis published the endosymbiotic theory in which she elegantly proposed a shared phylogenetic history between bacteria and mitochondria, both sharing the same ancestor [220]. Since then, this theory became the cornerstone of the modern molecular and cell biology.
The SCFAs, especially butyrate, were shown to enhance microbial diversity and promote the abundance the bacteria that secrete these compounds. On the other hands, they are also known to activate the AMP kinase, needed to promote mitochondrial biogenesis (see above). This remains a good example on how gut microbiota influence mitochondrial functions related to energy production, mitochondrial biogenesis, and ROS modulation. Moreover, bearing in mind that mitochondria are the predominant site of substrate oxidation and energy production, needed for the healthy muscular contraction, it is well-known that butyrate and urolithin A enhance skeletal muscle’s oxidative capacity and mitochondrial function. Hence, it is not surprising to say that metabolic health and mitochondrial health are synonymous, and these findings corroborate a muscle-microbiota re-inforcement in both directions [221]. In the same context, factors that regulate the microbiota quality and diversity like diet, probiotics, and others can have an effect on the muscular activity and resistance to stress in case of endurance exercise and chronic training of athletes [222].
In recent years, it has become increasingly evident that there is a bidirectional interaction between mitochondria and microbiota [223]. Even though we are still in the infancy of understanding such interplay, several reports revealed that specific microbiota composition (in terms of quality and diversity) is associated with many chronic inflammatory diseases, metabolic disorders, and even neuropsychological disorders (Fig. 4).
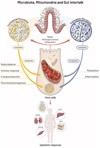 |
Figure 4 Scheme illustrating the crosstalk between microbiota and mitochondria and its systemic impact on the host |
Owing to the fact that mitochondria are the main source of ROS, it is obvious that these cellular powerhouses are key determinant of the innate immune response. Depending on the ROS level inside the cell, it can induce cell proliferation, pro-inflammatory cytokine release, or apoptosis [223]. Nonetheless, mitochondria invoke also adaptive immune responses thereby promoting adequate self-defenses against invaders such as pathogenic bacteria or viruses, e.g. the emerging COVID-19 virus [224].
Beside the mitochondrial role in regulation of healthy gut epithelial barrier [223] and in triggering chronic inflammatory response and cancer [225]. Jackson et al. reviewed the role of dysfunctional mitochondria in tumorigenesis. They explained how cancerous cells exhibit hybrid metabolism (both glycolysis and oxidative phosphorylation) in a trial of adapting to their microenvironment. Altered mitochondrial enzymes generate precursors needed for the fast cellular proliferation. They also mentioned how hypoxia drives the production of many oncometabolites and the progression of colorectal cancer. Many mtDNA mutations were also found in early neoplasia. The increased risk of colorectal cancer has been associated with dysbiosis of the gut microbiome and decreased production of SCFAs, but the exact involvement of microbiota in driving intestinal cancerogenesis is not fully understood [226].
In the context of gastrointestinal disorders, the effects of mitochondrial dysfunction and the microbiota dysbiosis on the development of inflammatory bowel diseases (IBD), such as Crohn’s disease (CD) and ulcerative colitis (UC) were also reported [225]. Particularly in the onset of CD, a recent study done by Mottawea et al. revealed that the onset of pediatric CD is associated with a significant decrease in butyrate-producing bacteria in the intestinal microbiota. As such, they argued that the depletion of butyrate-producing microbes from may diminish the capacity of H2S detoxification by the body, owing to the fact that butyrate activates the expression of the genes encoding the mitochondrial H2S detoxification components. This decrease in butyrate producers is associated with a significant increase in H2S-producing H2S, among which the Atopobium parvulum. Consequently, the enhanced production of the pro-inflammatory H2S caused additional metabolic stress and subsequently increased mucosal inflammation [227].
At a systemic level, the intriguing anti-inflammatory role of mitochondria is not surprising and had been associated with the maintenance of the systemic homeostasis of the body. Actually, dysfunctional mitochondria are well-known to activate many cytosolic multiprotein complexes that promote inflammation and cellular distress, referred to as inflammasomes (more about inflammasomes see below). Owing to the fact that some metabolites in the gut microbiota seem to interfere with mitochondrial oxidative stress and the activation of the inflammasomes, recent evidence and research showed that the gut microbiota can regulate crucial mitochondrial factors, coactivators, and enzymes involved in chronic metabolic disorders such as obesity and T2D [228]. Firstly, in obese patients, the altered adipose profile (accumulation of macronutrients in adipose tissues) stimulates the secretion of pro-inflammatory cytokines and contributes to ROS generation and oxidative stress which, in turn, trigger a systemic inflammation state with hyperinsulinemia and insulin resistance. In addition, oxidative stress has been showed to be closely related to the dysfunction of β-cell which are the central players in the insulin secretion. Of note, several papers reported that obese people present consistent changes in their gut microbiota as compared to normal individuals. However, and unfortunately, the molecular mechanisms by which the intestinal microbiota could contribute to the pathogenesis of such metabolic disorders are still poorly understood [228, 229].
The microbiota-mitochondria interplay was recently also assessed in the context of endometriosis. Classically, it was known that endometriosis is a local estrogen-mediated disorder upon retrograde menstruation. A study by George Anderson in 2019 showed that underlying mechanism of endometriosis is multifactorial including not only estrogen, but also mitochondrial dysregulation affected by immunity, gut and uterine microbiome, and vitamin A. Increased oxidative stress is evident in endometriosis and overly compensated by heightened levels of oxidative phosphorylation and ATP production. Moreover, the women with endometriosis showed an alteration in their gut, and their uterine microbiome [230]. The latter results are in concurrence with other results provides by another study in the same year by Ata et al. they conclude that the overall microbiome among women with endometriosis and normal individuals seems to be similar, but they showed many differences at the genus level of the bacteria. Further studies are needed to see if these differences are random and if the endometriosis can lead to microbiota dysbiosis or vice versa [231].
Finally the human microbiome is not only influenced by the chronic metabolic disorders but also by many therapeutic strategies such as the use of antibiotics, and dialysis in patients suffering from chronic kidney diseases. A pilot study conducted by one of us (M.E.) and his coworkers in 2018 was the first to reveal a change in the human microbiota associated with a mitochondrial dysfunction in hemodialysis (HD) patients (using citrate dialysate) and suffering from cramps. The authors explained the mitochondrial dysfunction in HD patients with cramps by an inadequate mitochondrial number or an alteration of many processes at the molecular levels among them dysfunction of their electron transport and ATP-synthesis-machinery. Nonetheless, they showed a change in the quality and diversity of the gut microbiota in HD patients with cramps compared to patients with no cramps. They attributed such dysbiosis to a modification of key factors and mediators produced by bacteria (butyrate, H2S, ROS, others), that, in turn, would alter the mitochondrial function and affect the skeletal muscular contractions [232]. The microbiome quality and diversity in patients with chronic kidney diseases undergoing hemodialysis and peritoneal diseases was also assessed and reviewed in the same year by Simões-Silva et al. [233]. In conclusion, considering that drug therapies could potentially interfere with the human microbiota and that in return, dysbiosis of the microbiota then can affect the severity of the disease’s symptoms as well as the host’s susceptibility to metabolic disorders such as obesity and diabetes [229], complementation of therapies with probiotics appears as advisable. Not only by the administration of pre-/probiotics, the manipulation of microbiota could also be achieved by fecal transplant, an appropriate diet, as well as phage therapy. Recently, Edeas and coworkers shed the light on the importance of phages not only for the elimination of pathogenic bacteria that lead to gastrointestinal infections but also for the modulation of the beneficial bacteria population by adding new functions such as metabolite biosynthesis (SCFAs and H2S) [234].
In summary, the cross-talk between mitochondria and microbiota plays a crucial role for maintaining homeostasis in the human body. The dysfunction of this bidirectional inter-talk, or one of its components, was shown to be implicated in the onset or the progression of many inflammatory disease states, cancers, metabolic, and neuropsychological disorders. In conclusion, targeting the mitochondria-microbiota interplay emerges as a whole new treatment strategy for many disorders and age-related morbidities. The challenge now is to find out how the microbiota in term of quality and diversity can be targeted via using different approaches which for example may include prebiotics, probiotics, special diets as well as phages.
Extracellular mitochondria in circulating blood
Human blood, the well-known opaque red liquid, is a circulating fluid supplying the body with needed oxygen and nutrients and removing metabolic waste products. It carries numerous cells and proteins suspended in it and it has vital functions for immunity (white blood cells or leucocytes, neutrophils, eosinophils), gas transport (red blood cells or erythrocytes), hemostasis (platelets and clotting factors) and the body’s electrolyte balance. It has been extensively studied for its important role in the pathophysiology of many metabolic disorders, auto-immune diseases, and cancers. Nevertheless, researchers most recently demonstrated that human blood which we thought to know so well surprisingly contains components that had not been detected previously. Dache et al. [235] and Song et al. [236] discovered the existence of cell-free circulating mitochondria in the bloodstream of humans and animals. These studies instantly suggest that plasma and any related products should be used with caution in any kind of application or research that might be affected by contamination with mitochondria. In the first study, the authors revealed the presence of highly stable structures containing whole mitochondrial genomes (between 200 000 and 3.7 million per mL of plasma) in circulating blood. Using fluorescence‐activated cell sorting analysis, fluorescence microscopy, and transmission electron microscopy to examine their size and density, as well as the integrity of their DNA, these particles were shown to be intact cell‐free mitochondria. Further functional studies (via Oxygen consumption analysis) done by the same team showed that these mitochondria seem to be “respiratory competent”, i.e. functional. However, the functionality of these cell-free mitochondria was re-evaluated by Antoine Stier and his group using high-resolution respirometry and they proposed that circulating cell-free mitochondria are unlikely to be functional in vivo mainly because they did not display any potential for oxidative phosphorylation [237].
The obvious question now is whether these “extracellular blood mitochondria” are just an accident or whether they may play a role in multiple pathologies including diabetes, immune and neurodegenerative disorders. Song et al. showed that purified mitochondria from bovine sera contribute to T- and B-cell activation, and human mitochondria upregulated the percentage of CD4+ and CD8+ T cells and thereby reduced the production of pro-inflammatory cytokines in blood. The authors suggested that the extracellular mitochondria in blood may function as a novel mediator for cell–cell communications and maintenance of homeostasis [236]. Results from another study by Yu et al. [238] revealed that platelet-derived mitochondria can trigger an up-regulation of naïve and central memory CD4+ T cells, the down-regulation of effector memory CD4+ T cells, and modulations of cytokine productions and gene expressions. Knowing that the auto-immune effector memory CD4+ T cells are key modulators of the initiation and development of type 1 diabetes (T1D) [239], the platelet-derived mitochondria emerge as novel and potential immune modulators to treat T1D and other autoimmune diseases. Other controversial pro- and anti-inflammatory roles for extracellular blood mitochondria were also discussed [240]. Interestingly, in the context of the role of these newly discovered extracellular blood mitochondria for human diseases, it was previously reported that the level of circulating cell-free mitochondrial DNA (ccf-mtDNA), distinct from the leukocyte mitochondrial DNA copy number, was elevated in patients suffering from major depressive disorder (MDD). The data shown suggested that plasma ccf-mtDNA may be reflective of certain aspects of MDD pathophysiology and of the response to some antidepressants [241]. Most recently even links between extracellular mtDNA and the SARS-CoV-2 coronavirus have been proposed. For example, Storci et al. showed that the increase in levels of the extracellular mitochondrial DNA is age- and sex-dependent and they suggest that this might explain why elderly men are more prone to hyper-inflammation and severe COVID complications than women [242]. For a more detailed discussion of a connection between mitochondria and the SARS-CoV-2 coronavirus, please see below.
Mitochondria and COVID-19
Currently, tremendous efforts are being undertaken to develop novel strategies for vaccination and therapies in order to cope with the Covid-19 pandemic. In addition, different ongoing research projects are dedicated to understanding the pathogenesis and the molecular mechanisms of the respiratory syndrome caused by the novel SARS-Cov-2 virus. However, one of the most important questions that is clinically asked and remains unfortunately not fully answered is why the elderly people, especially with co-morbidities are at higher risk of cytokine storm, sepsis, and multi-organ failure compared to younger populations.
Emerging evidence suggests that COVID-19 highjacks mitochondria of immune cells, replicates within mitochondrial structures, and impairs mitochondrial dynamics leading to cell death. Data suggest that mitochondria from COVID-19 infected cells are highly vulnerable, and vulnerability increases with age. Mitochondria are also known to interact with viral particles when they infect human host cells, engaging interferon and cytokine release, stimulating inflammation, and influencing viral survival and replication [243]. The novel SARS-CoV-2 uses its spike glycoprotein on the angiotensin-converting enzyme-2 (ACE-2) host receptor to enter human host cells via endocytosis [244]. Based on the fact that ACE-2 influences mitochondrial functions, Singh et al. argued that an age-related decline in ACE-2 expression and function correlates with decreased ATP production and altered activation of NADPH oxidase 4 in the mitochondria, which is normally used for ROS production [245]. The decrease in these components will prevent the cell from killing the virus and inhibit the infected cell apoptosis. Additionally, advanced studies showed that SARS-CoV-2 also influences mitochondrial function by acting on the estrogen-related receptor alpha, which is a nuclear receptor that regulates transcription of mitochondrial functions and energy homeostasis. It is intriguing that this hormonal regulation of mitochondrial targets and functions differs between men and women, which can partially explain why SARS-CoV-2 infection results in high rate of mortality in men compared to women [245, 246].
Once the SARS-Cov-2 enters the cell, the virus can directly manipulate the mitochondrial function to evade host cell immunity to its advantage and facilitate virus replication and COVID-19 disease onset. The viral RNA localizes the mitochondria and results in the mitochondrial DNA (mtDNA) release in the cytoplasm. This release will trigger the activation mtDNA-induced inflammasome and suppress innate and adaptive immunity [245]. Indeed, mtDNA itself acts as a danger-associated molecular pattern (DAMP). Its release in the cytoplasm, or into the extracellular compartment, results in a detrimental inflammatory response as suggested by Nakahira et al. who demonstrated the association of elevated cell-free, plasma mtDNA levels with increased ICU-related mortality [247].
In the same context, when severely damaged, the mitochondrial spinoffs released in the cytoplasm is accompanied by the upregulation of Ca2+ levels and the production of the pro-inflammatory cytokines, interleukin 6 (IL-6) and interleukin 1 beta (IL-1β) [248] which are the hallmarks of the Covid-19 cytokine storm [249]. Therefore, knowing that the mitochondrial dysfunction is associated with aging and age-related morbidities and metabolic syndromes, scientists now can comprehend why people who exhibit age- and comorbidities-related mitochondrial dysfunction have a poor prognosis in Covid-19 infection showing a high risk of sepsis, cytokine storm, and multi-organ failure [250]. Nonetheless, preventive treatments based on therapies improving mitochondrial turnover, dynamics and activity would be essential to protect against Covid-19 severity [250, 251].
On another note, it was suggested that mitochondrial disruption by SARS-Cov-2 could be also due to high ferritin levels, which by itself, leads to elevated iron-mediated oxidative stress, production of ROS, and the pro-inflammatory cytokine release [248, 252]. Additionally, the ferritin overload can disrupt glucose tolerance in these cells with mitochondrial oxidative stress, which has implications for diabetic patients [253]. Despite its strong association with high risk of cytokine storm and mortality in Covid-19 patients, the implication of hyper-ferritinemia in the progression of cases is not yet fully elucidated. However, as proposed by Edeas et al. it is crucial to investigate coexisting iron parameters in COVID-19 patients including transferrin saturation, plasma iron levels, non-transferrin bound iron (NTBI) as well as hepcidin. In a trial to target the iron homeostasis in COVID-19, the use of iron chelators, ferroptosis, and erythropotein could be a promising therapeutic approach, among others, that helps in the control of the disease progression [254].
Among the other factors that could exacerbate the severity of the Covid-19 infection via the sepsis and cytokine storm, it was believed that the gut microbiota and its dysbiosis possibly play a role in such an exacerbation. It was discussed by Vignesh et al. that the acute gastrointestinal inflammation in Covid-19 cases can lead to leaky gut allowing the flux of bacterial toxins and metabolites into the systemic circulation, worsening the septic state of patients [255]. Nonetheless, it was also demonstrated that respiratory viruses can lead to systemic infection trough lung-gut axis, especially with a poor intestinal integrity. This gut-respiratory link can explain why people exhibiting gastrointestinal symptoms of covid-19 are also prone to sepsis and cytokine storm [256].
Mitochondria and inflammation
Defined as the “Powerhouse of the cell” in 1957 [4] and as “Motor of cell death” in 1999 [257], a review sanctified mitochondria in 2017 as “Powerhouses of immunity” [12] indicating that this organelle also seems to lie at the heart of immunological responses. In 2011 it was demonstrated that inflammasome activity is activated by ROS derived from mitochondria [258]. Since then additional data were published demonstrating that “mitochondrial functions (are) being repurposed in unexpected ways to contribute to inflammatory signaling” [259]. For example, pro-or anti-inflammatory signals can be produced in mitochondria by modifying the level of Krebs cycle metabolites or by altering levels of ROS (briefly reviewed in [259]). In 2018 two papers came out demonstrating that the release of mitochondrial DNA or RNA to the cytoplasm can act as a signal triggering an antimicrobial defense response (reviewed in [259]). Zhong and colleagues showed that the engagement of Toll-like receptors may trigger the synthesis of new mtDNA which is then followed by oxidized mtDNA fragments leaving the organelle and activating inflammasomes [260]. Dhir et al. found that double-stranded RNA released from mitochondria into the cytosol can act as an antiviral signal [261]. These most recent discoveries about the role mitochondria play for immunological responses “could potentially be exploited for therapeutic gain in inflammation and cancer” [12].
Mitochondrial dysfunction in psychiatric illness
The term “schizophrenia” (Greek “splitting the mind”) was coined in 1908 by Eugen Bleuer (1857–1939), a Swiss psychiatrist [262]. About 50 years later, the first antipsychotic drug, chlorpromazine was introduced marking the beginning of serious biomedical research into the etiology of schizophrenia [263]. The involvement of impaired mitochondrial function in the pathophysiology of schizophrenia was hypothesized for the first time around at the beginnings of the 2000s, when Dorit Ben-Shachar proposed that “an abnormal cellular energy state can lead to alterations in neuronal function, plasticity and brain circuitry, and thereby to the cognitive and behavioral aberrations characteristic of schizophrenia” [264]. Although the exact mechanism by which mitochondria contribute to the pathophysiology of mental illness remains to be revealed, some data strongly supports mitochondrial involvement in etiology and pathophysiology of schizophrenia and bipolar disorder [265, 266]. According to a recent review focusing on brain energetics in animal models of depression, bipolar disorder, schizophrenia and autism, “most reported deficits included decreased activity in the electron transport chain, increased oxidative damage, decreased antioxidants defense, decreased ATP levels, and decreased mitochondrial potential” [267]. The further elucidation of the role mitochondria play in the pathogenesis of mental disorders will lead to a better understanding of therapeutic and side effects of psychotropics thereby improving current therapies for psychiatric patients [268]. For example, a recent study involving 48 schizophrenia and 27 bipolar disorder patients used a profile of mitochondrial parameters in lymphocytes to predict “the drug of choice” for individual patients. Strikingly, it was found that a long-term treatment with psychotropics normalized mitochondria-associated parameters in patients responding to therapy [269]. This “proof of concept study” conducted by Dorit Ben-Shachar and her colleagues opens a whole new avenue for the therapy of psychosis patients only 20 years after the same author for the first time predicted mitochondrial involvement in the pathophysiology of mental disorders.
Conclusions
Covering the entire burgeoning field of mitochondrial science in a single review, even in one composed of two parts, is difficult. We have tried emphasizing highlights in the development of mitochondrial medicine, tracking selected lines of research from their past to the present. We hope we have been successful first in communicating to the reader our fascination with this organelle and second in substantiating what we have written over a decade ago, that “the future of medicine will come through mitochondria” [13].
Abbreviations
AD: Alzheimer’s disease
AICAR: 5-aminoimdazole-4-carboxamide ribonucleoside
AMPK: AMP-Kinase
ATP: Adenosine triphosphate
CD: Crohn’s disease
cDNA: Complementary DNA
CL: Cardiolipin
CuZnSOD: Copper – Zinc Superoxide Dismutase
DED: Dry Eye Disease
DLC: Delocalized cation
ER: Endoplasmic reticulum
ETC: Electron transfer chain
FADH2: Reduced Flavin adenine dinucleotide
HD: Hemodialysis
IBD: Inflammatory bowel diseases
IMM: Inner mitochondrial membrane
KEAP/Nrf2: Kelch-like ECH-associated protein 1-nuclear factor (erythroid-derived 2)- like 2 regulatory pathway
MCU: Mitochondrial Calcium Uniporter
MFRTA: Mitochondrial Free Radical Theory of Aging
MICU1: Mitochondrial Calcium Uptake 1
mitoKATP: Mitochondrial inner membrane potassium channel
MnSOD: Manganese Superoxide Dismutase
MQC: Mitochondrial quality control
mtDNA: Mitochondrial DNA
mTOR: Mechanistic (or mammalian) target of rapamycin
MTPP: Methyl-triphenylphosphonium
NAD: Nicotinamide Adenine Dinucleotide
NADH2: Reduced Nicotinamide Adenine Dinucleotide
NCE: New Chemical Entity
NF-kB: Nuclear Factor binding near the k light chain gene in B cells
NRF: Nuclear Respiratory Factor
OXPHOS-ETC: Oxidative Phosphorylation Electron Transfer Chain
PGC-1: Peroxisome proliferator-activated receptor-gamma coactivator 1
PGC-1α: Peroxisome proliferator-activated receptor (PPAR)-ϫ coactivator-1α
PGC-1β: Peroxisome proliferator-activated receptor (PPAR)-ϫ coactivator-1β
PPARs: Peroxisome Proliferator-activated Receptors
PPAR-ϫ: Peroxisome proliferator-activated receptor gamma
QSAR: Quantitative structure-activity relations
RNS: Reactive Nitrogen Species
ROS: Reactive Oxygen Species
RTA 408: An investigational new drug (a synthetic triterpenoid)
SCFAs: Short chain fatty acids
SIRT: Sirtuin 1
UC: Ulcerative colitis
UCP: Uncoupling protein
UPR: Unfolded protein response
UPRmt: Mitochondrial unfolded protein response
VDAC: Voltage-Dependent Anion Channel
Conflict of interest
Volkmar Weissig is a Senior Board member at Life Sciences-Medicine of 4Open, published by EDP Sciences. Both authors did not take any action to influence the standard submission or peer-review process. The authors alone are responsible for the content and writing of this manuscript. This manuscript contains original material that has been previously published and is appropriately marked and cited. The authors have no conflicts of interest to declare.
Acknowledgments
The authors would like to thank all members of the World Mitochondrial Society (Paris, France) for their strong support of and for their active participation in our annual World Conference of Targeting Mitochondria series. Figures 1–3 were drawn by Mrs. Monica Henson (MWU Glendale).
References
- Weissig V, Edeas M (2021), Recent deveopments in mitochondrial medicine (part 1). 4Open 4, 1–13. https://doi.org/10.1051/fopen/2021002. [CrossRef] [Google Scholar]
- Tzagoloff A (1982), Mitochondria, Plenum Press, New York. ISBN 0-306-40799-X. [Google Scholar]
- Altmann R(1890), Die Elementarorganismen und ihre Beziehungen zu den Zellen, 1. Auflage, Von Veit & Comp Verlag, Leipzig. Deutsches Textarchiv. https://www.deutschestextarchiv.de/altmann_elementarorganismen_1890/9. Access: April 9, 2021. [Google Scholar]
- Siekevitz P (1957), Powerhouse of the cell. Scientific American 197, 131–144. [Google Scholar]
- Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B (1962), A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Invest 41(9), 1776–1804. https://doi.org/10.1172/JCI104637. PMCID: PMC291101; PMID: 14467237. [CrossRef] [PubMed] [Google Scholar]
- Luft R (1994), The development of mitochondrial medicine. Proc Natl Acad Sci U S A. 91(19), 8731–8738. https://doi.org/10.1073/pnas.91.19.8731. PMCID: PMC44681. [CrossRef] [PubMed] [Google Scholar]
- Nass S, Nass MM (1963), Intramitochondrial fibers with DNA characteristics. Ii. Enzymatic and other hydrolytic treatments. J Cell Biol 19, 613–629. PMID: 14086139. PMCID: PMC2106332. https://doi.org/10.1083/jcb.19.3.613 [CrossRef] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (1981), Sequence and organization of the human mitochondrial genome. Nature 290(5806), 457–465. https://doi.org/10.1038/290457a0. PMID: 7219534. [CrossRef] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Morgan-Hughes JA (1988), Deletions of muscle mitochondrial DNA. Nature 331(6158), 717–719. https://doi.org/10.1038/331717a0. PMID: 2830540. [CrossRef] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ 2nd, Nikoskelainen EK (1988), Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 242(4884), 1427–1430. https://doi.org/10.1126/science.3201231. PMID: 3201231. [CrossRef] [PubMed] [Google Scholar]
- Brown GC, Nicholls DG, Cooper CE (Eds.) (1999), Mitochondria and cell death, Princeton University Press. ISBN: 0-691-05026-0. [Google Scholar]
- Mills EL, Kelly B, O’Neill LAJ (2017), Mitochondria are the powerhouses of immunity. Nat Immunol 18(5), 488–498. https://doi.org/10.1038/ni.3704. PMID: 28418387. [CrossRef] [PubMed] [Google Scholar]
- Edeas M, Weissig V (2013), Targeting mitochondria: strategies, innovations and challenges: the future of medicine will come through mitochondria. Mitochondrion 13(5), 389–390. https://doi.org/10.1016/j.mito.2013.03.009. [CrossRef] [PubMed] [Google Scholar]
- De Benedictis G, Franceschi C (2006), The unusual genetics of human longevity. Sci Aging Knowl Environ 2006(10), pe20. https://doi.org/10.1126/sageke.2006.10.pe20. PMID: 16807484. [CrossRef] [Google Scholar]
- Salmon AB, Richardson A, Pérez VI (2010), Update on the oxidative stress theory of aging: does oxidative stress play a role in aging or healthy aging? Free Radic Biol Med 48(5), 642–655. https://doi.org/10.1016/j.freeradbiomed.2009.12.015. PMID: 20036736. [CrossRef] [PubMed] [Google Scholar]
- Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G, Cacciatore F, Bonaduce D, Abete P (2018), Oxidative stress, aging, and diseases. Clin Interv Aging 13, 757–772. https://doi.org/10.2147/CIA.S158513. eCollection 2018. PMID: 29731617. [CrossRef] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013), The hallmarks of aging. Cell 153(6), 1194–1217. https://doi.org/10.1016/j.cell.2013.05.039. PMID: 23746838. [CrossRef] [PubMed] [Google Scholar]
- Harman D (1956), Aging: a theory based on free radical and radiation chemistry. J Gerontol 11(3), 298–300. https://doi.org/10.1093/geronj/11.3.298. PMID: 13332224. [CrossRef] [PubMed] [Google Scholar]
- Harman D (1972), The biologic clock: the mitochondria? J Am Geriatr Soc 20(4), 145–147. https://doi.org/10.1111/j.1532-5415.1972.tb00787.x. PMID: 5016631. [CrossRef] [PubMed] [Google Scholar]
- Miquel J, Economos AC, Fleming J, Johnson JE Jr (1980), Mitochondrial role in cell aging. Exp Gerontol 15(6), 575–591. https://doi.org/10.1016/0531-5565(80)90010-8. PMID: 7009178. [CrossRef] [PubMed] [Google Scholar]
- Sastre J, Pallardó FV, Plá R, Pellín A, Juan G, O’Connor JE, Estrela JM, Miquel J, Viña J (1996), Aging of the liver: age-associated mitochondrial damage in intact hepatocytes. Hepatology 24(5), 1199–1205. https://doi.org/10.1002/hep.510240536. PMID: 8903398. [CrossRef] [PubMed] [Google Scholar]
- Hagen TM, Yowe DL, Bartholomew JC, Wehr CM, Do KL, Park JY, Ames BN (1997), Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc Natl Acad Sci U S A 94(7), 3064–3069. https://doi.org/10.1073/pnas.94.7.3064. PMID: 9096346. [CrossRef] [PubMed] [Google Scholar]
- Nass MM, Nass S (1963), Intramitochondrial fibers with DNA characteristics. I. Fixation and electron staining reactions. J Cell Biol 19(3), 593–611. https://doi.org/10.1083/jcb.19.3.593. PMID: 14086138 PMCID: PMC2106331. [CrossRef] [PubMed] [Google Scholar]
- Wallace DC (1992), Diseases of the mitochondrial DNA. Annu Rev Biochem 61, 1175–1212. https://doi.org/10.1146/annurev.bi.61.070192.005523. PMID: 1497308. [CrossRef] [PubMed] [Google Scholar]
- Cooper JM, Mann VM, Schapira AH (1992), Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: effect of ageing. J Neurol Sci 113(1), 91–98. https://doi.org/10.1016/0022-510x(92)90270-u. PMID: 1469460. [CrossRef] [PubMed] [Google Scholar]
- Hattori K, Tanaka M, Sugiyama S, Obayashi T, Ito T, Satake T, Hanaki Y, Asai J, Nagano M, Ozawa T (1991), Age-dependent increase in deleted mitochondrial DNA in the human heart: possible contributory factor to presbycardia. Am Heart J 121(6 Pt 1), 1735–1742. https://doi.org/10.1016/0002-8703(91)90020-i. PMID: 2035386. [CrossRef] [PubMed] [Google Scholar]
- Sun J, Folk D, Bradley TJ, Tower J (2002), Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics 161(2), 661–672. https://doi.org/10.1093/genetics/161.2.661. PMID: 12072463. [CrossRef] [PubMed] [Google Scholar]
- Orr WC, Sohal RS (1994), Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 263(5150), 1128–1130. https://doi.org/10.1126/science.8108730. PMID: 8108730. [CrossRef] [PubMed] [Google Scholar]
- Huang TT, Carlson EJ, Gillespie AM, Shi Y, Epstein CJ (2000), Ubiquitous overexpression of CuZn superoxide dismutase does not extend life span in mice. J Gerontol A Biol Sci Med Sci 55(1), B5–B9. https://doi.org/10.1093/gerona/55.1.b5. PMID: 10719757. [CrossRef] [PubMed] [Google Scholar]
- Simmons TW, Jamall IS (1989), Relative importance of intracellualr glutathione peroxidase and catalasein vivo for prevention of peroxidation to the heart. Cardiovasc Res 23(9), 774–779. PMID: 2611816. https://doi.org/10.1093/cvr/23.9.774. [CrossRef] [PubMed] [Google Scholar]
- Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang TT, Nelson J, Strong R, Richardson A (2003), Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genom 16(1), 29–37. https://doi.org/10.1152/physiolgenomics.00122.2003. PMID: 14679299. [CrossRef] [PubMed] [Google Scholar]
- Andziak B, O’Connor TP, Qi W, DeWaal EM, Pierce A, Chaudhuri AR, Van Remmen H, Buffenstein R (2006), High oxidative damage levels in the longest-living rodent, the naked mole-rat. Aging Cell 5(6), 463–471. https://doi.org/10.1111/j.1474-9726.2006.00237.x. PMID: 17054663. [CrossRef] [PubMed] [Google Scholar]
- Richardson A (2012), Is the free radical (oxidative stress) theory of aging dead? Gerontologist 52, 405–405. [Google Scholar]
- Pérez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y, Richardson A (2009), Is the oxidative stress theory of aging dead? Biochim Biophys Acta 1790(10), 1005–1014. https://doi.org/10.1016/j.bbagen.2009.06.003. PMID: 19524016. [CrossRef] [PubMed] [Google Scholar]
- Sharma A, Smith HJ, Yao P, Mair WB (2019), Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep 20(12), e48395. https://doi.org/10.15252/embr.201948395. PMID: 31667999; PMCID: PMC6893295. [CrossRef] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K, Nishimoto I (2001), A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc Natl Acad Sci U S A 98(11), 6336–6341. https://doi.org/10.1073/pnas.101133498. PMID: 11371646. [CrossRef] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Ito Y, Sudo H, Hata M, Arakawa E, Abe Y, Kita Y, Nishimoto I (2001), Detailed characterization of neuroprotection by a rescue factor humanin against various Alzheimer’s disease-relevant insults. J Neurosci 21(23), 9235–9245. https://doi.org/10.1523/JNEUROSCI.21-23-09235.2001. PMID: 11717357; PMCID: PMC6763898. [CrossRef] [PubMed] [Google Scholar]
- Hashimoto Y, Ito Y, Niikura T, Shao Z, Hata M, Oyama F, Nishimoto I (2001), Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein. Biochem Biophys Res Commun 283(2) 460–468. https://doi.org/10.1006/bbrc.2001.4765. PMID: 11327724. [CrossRef] [PubMed] [Google Scholar]
- Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AMJ, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, Filipovska A, Mattick JS (2011), The human mitochondrial transcriptome. Cell 146(4), 645–658. https://doi.org/10.1016/j.cell.2011.06.051. PMID: 21854988; PMCID: PMC3160626. [CrossRef] [PubMed] [Google Scholar]
- Mercer TR, Wilhelm D, Dinger ME, Soldà G, Korbie DJ, Glazov EA, Truong V, Schwenke M, Simons C, Matthaei KI, Saint R, Koopman P, Mattick JS (2011), Expression of distinct RNAs from 3’ untranslated regions. Nucleic Acids Res 39(6), 2393–2403. https://doi.org/10.1093/nar/gkq1158. PMID: 21075793; PMCID: PMC3064787. [CrossRef] [PubMed] [Google Scholar]
- Merry TL, Chan A, Woodhead JST, Reynolds JC, Kumagai H, Kim SJ, Lee C (2020), Mitochondrial-derived peptides in energy metabolism. Am J Physiol Endocrinol Metab 319(4), E659–E666. https://doi.org/10.1152/ajpendo.00249.2020. PMID: 32776825; PMCID: PMC7750512. [CrossRef] [PubMed] [Google Scholar]
- Yen K, Mehta HH, Kim SJ, Lue YH, Hoang J, Guerrero N, Port J, Bi Q, Navarrete G, Brandhorst S, Lewis KN, Wan J, Swerdloff R, Mattison JA, Buffenstein R, Breton CV, Wang C, Longo V, Atzmon G, Wallace D, Barzilai N, Cohen P (2020), The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan. Aging (Albany NY) 12(12), 11185–11199. https://doi.org/10.18632/aging.103534. PMID: 32575074. [CrossRef] [PubMed] [Google Scholar]
- Berry BJ, Kaeberlein M (2021), An energetics perspective on geroscience: mitochondrial protonmotive force and aging. Geroscience. https://doi.org/10.1007/s11357-021-00365-7. PMID: 33864592. [PubMed] [Google Scholar]
- Duchen MR(2010), Mitochondrial function and dysfunction in human disease – potential therapeutic targets from cradle to grave, in: M. Edeas(Ed.), 1st World Conference on Targeting Mitochondria, ISANH, p. 22. [Google Scholar]
- Duchen MR, Szabadkai G (2010), Roles of mitochondria in human disease. Essays Biochem 47, 115–137. https://doi.org/10.1042/bse0470115. PMID: 20533904. [CrossRef] [PubMed] [Google Scholar]
- Qiu Z, Wei Y, Song Q, Du B, Wang H, Chu Y, Hu Y (2019), The role of myocardial mitochondrial quality control in heart failure. Front Pharmacol 10, 1404. https://doi.org/10.3389/fphar.2019.01404. PMID: 31866862. [CrossRef] [PubMed] [Google Scholar]
- Stotland A, Gottlieb RA (2015), Mitochondrial quality control: easy come, easy go. Biochim Biophys Acta 1853(10 Pt B), 2802–2811. https://doi.org/10.1016/j.bbamcr.2014.12.041. PMID: 25596427. [CrossRef] [PubMed] [Google Scholar]
- Popov LD (2020), Mitochondrial biogenesis: an update. J Cell Mol Med 24(9), 4892–4899. https://doi.org/10.1111/jcmm.15194. PMID: 32279443. [CrossRef] [PubMed] [Google Scholar]
- Chan DC (2020), Mitochondrial dynamics and its involvement in disease. Review Annu Rev Pathol 15, 235–259. https://doi.org/10.1146/annurev-pathmechdis-012419-032711. PMID: 31585519. [CrossRef] [PubMed] [Google Scholar]
- Bakula D, Scheibye-Knudsen M (2020), MitophAging: mitophagy in aging and disease. Front Cell Dev Biol 8, 239. https://doi.org/10.3389/fcell.2020.00239. PMID: 32373609. [CrossRef] [PubMed] [Google Scholar]
- Leuenberger D, Curran SP, Koehler CM(2005), Mitochondrial biogenesis, in: C. Mullins (Ed.), The biogenesis of cellular organelles, Springer, pp. 138–163. ISBN 978-0- 387-26867-5. [CrossRef] [Google Scholar]
- Holloszy JO (1967), Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 242(9), 2278–2282. PMID: 4290225. [CrossRef] [PubMed] [Google Scholar]
- Holloszy JO (2011), Regulation of mitochondrial biogenesis and GLUT4 expression by exercise. Compr Physiol 1(2), 921–940. https://doi.org/10.1002/cphy.c100052. PMID: 23737207. [CrossRef] [PubMed] [Google Scholar]
- Hagberg JM, Coyle EF, Baldwin KM, Cartee GD, Fontana L, Joyner MJ, Kirwan JP, Seals DR, Weiss EP (2019), The historical context and scientific legacy of John O. Holloszy. J Appl Physiol (1985) 127(2), 277–305. https://doi.org/10.1152/japplphysiol.00669.2018. PMID: 30730811. [CrossRef] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM (1998), A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92(6), 829–839. https://doi.org/10.1016/s0092-8674(00)81410-5. PMID: 9529258. [CrossRef] [PubMed] [Google Scholar]
- Valero T (2014), Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des 20(35), 5507–5509. https://doi.org/10.2174/138161282035140911142118. PMID: 24606795. [CrossRef] [PubMed] [Google Scholar]
- Martinez-Redondo V, Pettersson AT, Ruas JL (2015), The hitchhiker’s guide to PGC-1alpha isoform structure and biological functions. Diabetologia 58(9), 1969–1977. https://doi.org/10.1007/s00125-015-3671-z. PMID: 26109214. [CrossRef] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, Kelly DP (2012), Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23(9), 459–466. https://doi.org/10.1016/j.tem.2012.06.006. PMID: 22817841. [CrossRef] [PubMed] [Google Scholar]
- Wenz T (2013), Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion 13(2), 134–142. https://doi.org/10.1016/j.mito.2013.01.006. PMID: 23347985. [CrossRef] [PubMed] [Google Scholar]
- Komen JC, Thorburn DR (2014), Turn up the power – pharmacological activation of mitochondrial biogenesis in mouse models. Review Br J Pharmacol 171(8), 1818–1836. https://doi.org/10.1111/bph.12413. PMID: 24102298. [CrossRef] [Google Scholar]
- Kressler D, Schreiber SN, Knutti D, Kralli A (2002), The PGC-1-related protein PERC is a selective coactivator of estrogen receptor alpha. J Biol Chem 277(16), 13918–13925. https://doi.org/10.1074/jbc.M201134200. PMID: 11854298. [CrossRef] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM (2005), Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab 1(6), 361–370. https://doi.org/10.1016/j.cmet.2005.05.004. PMID: 16054085. [CrossRef] [PubMed] [Google Scholar]
- Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP (2008), Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev 22(14), 1948–1961. https://doi.org/10.1101/gad.1661708. PMID: 18628400. [CrossRef] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jäger S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM (2004), Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119(1), 121–135. https://doi.org/10.1016/j.cell.2004.09.013. PMID: 15454086. [CrossRef] [PubMed] [Google Scholar]
- Jornayvaz FR, Shulman GI (2010), Regulation of mitochondrial biogenesis. Essays Biochem 47, 69–84. https://doi.org/10.1042/bse0470069. PMID: 20533901. [CrossRef] [PubMed] [Google Scholar]
- Johri A, Chandra A, Beal MF (2013), PGC-1alpha, mitochondrial dysfunction, and Huntington’s disease. Free Radic Biol Med 62, 37–46. https://doi.org/10.1016/j.freeradbiomed.2013.04.016. PMID: 23602910. [CrossRef] [PubMed] [Google Scholar]
- Scarpulla RC (2011), Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813 7, 1269–1278. https://doi.org/10.1016/j.bbamcr.2010.09.019. PMID: 20933024. [Google Scholar]
- Fernandez-Marcos PJ, Auwerx J (2011), Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93(4), 884S–890S. https://doi.org/10.3945/ajcn.110.001917. PMID: 21289221. [CrossRef] [PubMed] [Google Scholar]
- Scarpulla RC (2012), Nucleus-encoded regulators of mitochondrial function: integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim Biophys Acta 1819 9–10, 1088–1097. https://doi.org/10.1016/j.bbagrm.2011.10.011. PMID: 22080153. [Google Scholar]
- Hofer A, Noe N, Tischner C, Kladt N, Lellek V, Schauß A, Wenz T (2014), Defining the action spectrum of potential PGC-1alpha activators on a mitochondrial and cellular level in vivo. Hum Mol Genet 23(9), 2400–2415. https://doi.org/10.1093/hmg/ddt631. PMID: 24334768. [CrossRef] [PubMed] [Google Scholar]
- Galinanes M, Bullough D, Mullane KM, Hearse DJ (1992), Sustained protection by acadesine against ischemia- and reperfusion-induced injury. Studies in the transplanted rat heart. Circulation 86(2), 589–597. https://doi.org/10.1161/01.cir.86.2.589. PMID: 1638724. [CrossRef] [PubMed] [Google Scholar]
- Thomas A, Beuck S, Eickhoff JC, Guddat S, Krug O, Kamber M, Schänzer W, Thevis M (2010), Quantification of urinary AICAR concentrations as a matter of doping controls. Anal Bioanal Chem 396(8), 2899–2908. https://doi.org/10.1007/s00216-010-3560-8. PMID: 20225061. [CrossRef] [PubMed] [Google Scholar]
- Thevis M, Sigmund G, Geyer H, Schänzer W (2010), Stimulants and doping in sport. Endocrinol Metab Clin North Am 39(1), 89–105, ix. https://doi.org/10.1016/j.ecl.2009.10.011. PMID: 20122452. [CrossRef] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Baird L, Holmström KM, Meyer CJ, Abramov AY (2015), The spatiotemporal regulation of the Keap1-Nrf2 pathway and its importance in cellular bioenergetics. Biochem Soc Trans 43(4), 602–610. https://doi.org/10.1042/BST20150003. PMID: 26551700. [CrossRef] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Abramov AY (2015), The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med 88(Pt B), 179–188. https://doi.org/10.1016/j.freeradbiomed.2015.04.036. PMID: 25975984. [CrossRef] [PubMed] [Google Scholar]
- Pall ML, Levine S (2015), Nrf2, a master regulator of detoxification and also antioxidant, anti-inflammatory and other cytoprotective mechanisms, is raised by health promoting factors. Sheng Li Xue Bao 67(1), 1–18. PMID: 25672622. [PubMed] [Google Scholar]
- Weissig V (2020), Drug development for the therapy of mitochondrial diseases. Trends Mol Med 26(1), 40–57. https://doi.org/10.1016/j.molmed.2019.09.002. PMID: 31727544. [Google Scholar]
- Han X, Nonaka K, Kato H, Yamaza H, Sato H, Kifune T, Hirofuji Y, Masuda K (2019), Osteoblastic differentiation improved by bezafibrate-induced mitochondrial biogenesis in deciduous tooth-derived pulp stem cells from a child with Leigh syndrome. Biochem Biophys Rep 17, 32–37. https://doi.org/10.1016/j.bbrep.2018.11.003. PMID: 30533535. [PubMed] [Google Scholar]
- Lewis MR, Lewis WH (1914), Mitochondria in tissue culture. Science 39(1000), 330–333. https://doi.org/10.1126/science.39.1000.330. PMID: 17794648. [CrossRef] [PubMed] [Google Scholar]
- Lewis MR, Lewis WH (1915), Mitochondria (and other cytoplasmic structures) in tissue cultures. Am J Anat 17, 339–401. https://doi.org/10.1002/aja.1000170304. [CrossRef] [Google Scholar]
- Chan DC (2006), Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22, 79–99. https://doi.org/10.1146/annurev.cellbio.22.010305.104638. PMID: 16704336. [CrossRef] [PubMed] [Google Scholar]
- Chen H, Chan DC (2005), Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet 14(2), R283–R289. https://doi.org/10.1093/hmg/ddi270. PMID: 16244327. [CrossRef] [PubMed] [Google Scholar]
- Griffin EE, Graumann J, Chan DC (2005), The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol 170(2), 237–248. https://doi.org/10.1083/jcb.200503148. PMID: 16009724. [CrossRef] [PubMed] [Google Scholar]
- Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA (2006), A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol 8(11), 1255–1262. https://doi.org/10.1038/ncb1487. PMID: 17028579. [CrossRef] [PubMed] [Google Scholar]
- Chan DC (2006), Dissecting mitochondrial fusion. Dev Cell 11(5), 592–594. https://doi.org/10.1016/j.devcel.2006.10.009. PMID: 17084350. [CrossRef] [PubMed] [Google Scholar]
- Chan DC (2006), Mitochondria: dynamic organelles in disease, aging, and development. Cell 125(7), 1241–1252. https://doi.org/10.1016/j.cell.2006.06.010. PMID: 16814712. [CrossRef] [PubMed] [Google Scholar]
- Chan DC (2012), Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46, 265–287. https://doi.org/10.1146/annurev-genet-110410-132529. PMID: 22934639. [CrossRef] [PubMed] [Google Scholar]
- Yu R, Lendahl U, Nistér M, Zhao J (2020), Regulation of mammalian mitochondrial dynamics: opportunities and challenges. Front Endocrinol (Lausanne) 11, 374. https://doi.org/10.3389/fendo.2020.00374. PMID: 32595603. [CrossRef] [PubMed] [Google Scholar]
- De Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F (1955), Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J 60(4), 604–617. https://doi.org/10.1042/bj0600604. PMID: 13249955. [CrossRef] [PubMed] [Google Scholar]
- Berthet J, Appelmans F, De Duve C (1951), Tissue fractionation studies. II. The nature of the linkage between acid phosphatase and mitochondria in rat-liver tissue. Biochem J 50(2), 182–189. https://doi.org/10.1042/bj0500182. PMID: 14904390. [CrossRef] [PubMed] [Google Scholar]
- Appelmans F, Wattiaux R, Duve De (1955), Tissue fractionation studies. 5. The association of acid phosphatase with a special class of cytoplasmic granules in rat liver. Biochem J 59(3), 438–445. https://doi.org/10.1042/bj0590438. PMID: 14363114. [CrossRef] [PubMed] [Google Scholar]
- De Duve C (2005), The lysosome turns fifty. Nat Cell Biol 7(9), 847–849. https://doi.org/10.1038/ncb0905-847. PMID: 16136179. [CrossRef] [PubMed] [Google Scholar]
- Ashford TP, Porter KR (1962), Cytoplasmic components in hepatic cell lysosomes. J Cell Biol 12(1), 198–202. https://doi.org/10.1083/jcb.12.1.198. PMID: 13862833. [CrossRef] [PubMed] [Google Scholar]
- Deter RL, De Duve C (1967), Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol 33(2), 437–449. https://doi.org/10.1083/jcb.33.2.437. PMID: 4292315. [CrossRef] [PubMed] [Google Scholar]
- Deter RL, Baudhuin P, De Duve C (1967), Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol 35(2), C11–C16. https://doi.org/10.1083/jcb.35.2.c11. PMID: 6055998. [CrossRef] [PubMed] [Google Scholar]
- Camougrand N, Kissová I, Velours G, Manon S (2004), Uth1p: a yeast mitochondrial protein at the crossroads of stress, degradation and cell death. FEMS Yeast Res 5(2), 133–140. https://doi.org/10.1016/j.femsyr.2004.05.001. PMID: 15489196. [CrossRef] [PubMed] [Google Scholar]
- Kissova I, Deffieu M, Manon S, Camougrand N (2004), Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem 279(37), 39068–39074. https://doi.org/10.1074/jbc.M406960200. PMID: 15247238. [CrossRef] [PubMed] [Google Scholar]
- Lemasters JJ (2005), Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8(1), 3–5. https://doi.org/0.1089/rej.2005.8.3. PMID: 15798367. [Google Scholar]
- Saxton RA, Sabatini DM (2017), mTOR signaling in growth, metabolism, and disease. Cell 169(2), 361–371. https://doi.org/10.1016/j.cell.2017.03.035. PMID: 28388417. [CrossRef] [PubMed] [Google Scholar]
- Vezina C, Kudelski A, Sehgal SN (1975), Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 28(10), 721–726. https://doi.org/10.7164/antibiotics.28.721. PMID: 1102508. [CrossRef] [PubMed] [Google Scholar]
- Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT (1995), Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem 270(2), 815–822. https://doi.org/10.1074/jbc.270.2.815. PMID: 7822316. [PubMed] [Google Scholar]
- Wiederrecht GJ, Sabers CJ, Brunn GJ, Martin MM, Dumont FJ, Abraham RT (1995), Mechanism of action of rapamycin: new insights into the regulation of G1-phase progression in eukaryotic cells. Prog Cell Cycle Res 1, 53–71. https://doi.org/10.1007/978-1-4615-1809-9_5. PMID: 9552353. [CrossRef] [PubMed] [Google Scholar]
- Lipton JO, Sahin M (2014), The neurology of mTOR. Neuron 84(2), 275–291. https://doi.org/10.1016/j.neuron.2014.09.034. PMID: 25374355. [CrossRef] [PubMed] [Google Scholar]
- Hay N, Sonenberg N (2004), Upstream and downstream of mTOR. Genes Dev 18(16), 1926–1945. https://doi.org/10.1101/gad.1212704. PMID: 15314020; PMCID: PMC7160495. [CrossRef] [PubMed] [Google Scholar]
- Kaufman RJ (2002), Orchestrating the unfolded protein response in health and disease. J Clin Invest 110(10), 1389–1398. https://doi.org/10.1172/JCI16886. PMID: 12438434. [CrossRef] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM (2001), The unfolding tale of the unfolded protein response. Cell 107(7), 827–830. https://doi.org/10.1016/s0092-8674(01)00623-7. PMID: 11779459. [CrossRef] [PubMed] [Google Scholar]
- Gething MJ, Sambrook J (1992), Protein folding in the cell. Nature 355(6355), 33–45. https://doi.org/10.1038/355033a0. PMID: 1731198. [CrossRef] [PubMed] [Google Scholar]
- Srere PA, Sumegi B (1986), Organization of the mitochondrial matrix. Adv Exp Med Biol 194, 13–25. https://doi.org/10.1007/978-1-4684-5107-8_2. PMID: 3529854. [CrossRef] [PubMed] [Google Scholar]
- Hoogenraad N (2017), A brief history of the discovery of the mitochondrial unfolded protein response in mammalian cells. J Bioenerg Biomembr 49(4), 293–295. https://doi.org/10.1007/s10863-017-9703-2. PMID: 28429159. [CrossRef] [PubMed] [Google Scholar]
- Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Høj BP, Hoogenraad NJ (1996), Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem 240(1), 98–103. https://doi.org/10.1111/j.1432-1033.1996.0098h.x. PMID: 8797841. [CrossRef] [PubMed] [Google Scholar]
- Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, Hoogenraad N, Sans M, Sartor RB, Haller D (2012), Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 61(9), 1269–1278. https://doi.org/10.1136/gutjnl-2011-300767. PMID: 21997551. [CrossRef] [PubMed] [Google Scholar]
- Ryan MT, Herd SM, Sberna G, Samuel MM, Hoogenraad NJ, Høj PB (1997), The genes encoding mammalian chaperonin 60 and chaperonin 10 are linked head-to-head and share a bidirectional promoter. Gene 196(1–2), 9–17. https://doi.org/10.1016/s0378-1119(97)00111-x. PMID: 9322735. [CrossRef] [PubMed] [Google Scholar]
- Ryan MT, Naylor DJ, Høj PB, Clark MS, Hoogenraad NJ (1997), The role of molecular chaperones in mitochondrial protein import and folding. Int Rev Cytol 174, 127–193. https://doi.org/10.1016/s0074-7696(08)62117-8. PMID: 9161007. [CrossRef] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ (2002), A mitochondrial specific stress response in mammalian cells. EMBO J 21(17), 4411–4419. https://doi.org/10.1093/emboj/cdf445. PMID: 12198143. [CrossRef] [PubMed] [Google Scholar]
- Smyrnias I, Gray SP, Okonko DO, Sawyer G, Zoccarato A, Catibog N, López B, González A, Ravassa S, Díez J, Shah AM (2019), Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J Am Coll Cardiol 73(14), 1795–1806. https://doi.org/10.1016/j.jacc.2018.12.087. PMID: 30975297. [CrossRef] [PubMed] [Google Scholar]
- Siegelin MD, Dohi T, Raskett CM, Orlowski GM, Powers CM, Gilbert CA, Ross AH, Plescia J, Altieri DC (2011), Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clin Invest 121(4), 1349–1360. https://doi.org/10.1172/JCI44855. PMID: 21364280. [CrossRef] [PubMed] [Google Scholar]
- Sen R, Baltimore D (1986), Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46(5), 705–716. https://doi.org/10.1016/0092-8674(86)90346-6. PMID: 3091258. [CrossRef] [PubMed] [Google Scholar]
- Zhang Q, Lenardo MJ, Baltimore D (2017), 30 years of NF-kappa B: a blossoming of relevance to human pathobiology. Cell 168(1–2), 37–57. https://doi.org/10.1016/j.cell.2016.12.012. PMID: 28086098. [CrossRef] [PubMed] [Google Scholar]
- Smale ST (2012), Dimer-specific regulatory mechanisms within the NF-kappa B family of transcription factors. Immunol Rev 246(1), 193–204. https://doi.org/10.1111/j.1600-065X.2011.01091.x. PMID: 22435556. [CrossRef] [PubMed] [Google Scholar]
- Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, Ragoussis J, Udalova IA, Smale ST, Bulyk ML (2012), Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappa B family DNA binding. Nat Immunol 13(1), 95–102. https://doi.org/10.1038/ni.2151. PMID: 22101729. [CrossRef] [Google Scholar]
- Williams LM, Gilmore TD (2020), Looking down on NF-kappa B. Mol Cell Biol 40(15), e00104–e00120. https://doi.org/10.1128/MCB.00104-20. PMID: 32393609. [CrossRef] [PubMed] [Google Scholar]
- Ivanenkov YA, Balakin KV, Lavrovsky Y (2011), Small molecule inhibitors of NF-kB and JAK/STAT signal transduction pathways as promising anti-inflammatory therapeutics. Mini Rev Med Chem 11(1), 55–78. https://doi.org/10.2174/138955711793564079. PMID: 21034406. [CrossRef] [PubMed] [Google Scholar]
- Mattson MP, Culmsee C, Yu Z, Camandola S (2000), Roles of nuclear factor kappa B in neuronal survival and plasticity. J Neurochem 74(2), 443–456. https://doi.org/10.1046/j.1471-4159.2000.740443.x. PMID: 10646495. [Google Scholar]
- Albensi BC (2019), What is nuclear factor kappa B (NF-kappa B) doing in and to the mitochondrion? Front Cell Dev Biol 7, 154. https://doi.org/10.3389/fcell.2019.00154. eCollection 2019. PMID: 31448275. [CrossRef] [PubMed] [Google Scholar]
- Bottero V, Busuttil V, Loubat A, Magné N, Fischel JL, Milano G, Peyron JF (2001), Activation of nuclear factor kappa B through the IKK complex by the topoisomerase poisons SN38 and doxorubicin: a brake to apoptosis in HeLa human carcinoma cells. Cancer Res 61(21), 7785–7791. PMID: 11691793. [PubMed] [Google Scholar]
- Bottero V, Rossi F, Samson M, Mari M, Hofman P, Peyron JF (2001), I kappa B-alpha, the NF-kappa B inhibitors subunit, interacts with ANT, the mitochondrial ATP/ADP translocator. J Biol Chem 276(24), 21317–21324. https://doi.org/10.1074/jbc.M005850200. PMID: 11287411. [CrossRef] [PubMed] [Google Scholar]
- Cogswell PC, Kashatus DF, Keifer JA, Guttridge DC, Reuther JY, Bristow C, Roy S, Nicholson DW, Baldwin AS Jr (2003), NF-kappa B and I kappa B alpha are found in the mitochondria – evidence for regulation of mitochondrial gene expression by NF-kappa B. J Biol Chem 278(5), 2963–2968. https://doi.org/10.1074/jbc.M209995200. PMID: 12433922. [CrossRef] [PubMed] [Google Scholar]
- Guseva NV, Taghiyev AF, Sturm MT, Rokhlin OW, Cohen MB (2004), Tumor necrosis factor-related apoptosis-inducing ligand-mediated activation of mitochondria-associated nuclear factor-kappa B in prostatic carcinoma cell lines. Mol Cancer Res 2(10), 574–584. PMID: 15498932. [PubMed] [Google Scholar]
- Zamora M, Meroño C, Viñas O, Mampel T (2004), Recruitment of NF-kappa B into mitochondria is involved in adenine nucleotide translocase 1 (ANT1)-induced apoptosis. J Biol Chem 279(37), 38415–38423. https://doi.org/10.1074/jbc.M404928200. PMID: 15231833. [CrossRef] [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, Kan YW (1994), Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA 91(21), 9926–9930. https://doi.org/10.1073/pnas.91.21.9926. PMID: 7937919. [CrossRef] [PubMed] [Google Scholar]
- Chan K, Lu R, Chang JC, Kan YW (1996), NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A 93(24), 13943–13948. https://doi.org/10.1073/pnas.93.24.13943. PMID: 8943040. [CrossRef] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y (1997), An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236(2), 313–322. https://doi.org/10.1006/bbrc.1997.6943. PMID: 9240432. [CrossRef] [PubMed] [Google Scholar]
- Hayes JD, Dinkova-Kostova AT (2014), The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39(4), 199–218. https://doi.org/10.1016/j.tibs.2014.02.002. PMID: 24647116. [CrossRef] [PubMed] [Google Scholar]
- Yamamoto M, Kensler TW, Motohashi H (2018), The KEAP1-NRF2 System: a thiol- based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 98(3), 1169–1203. https://doi.org/10.1152/physrev.00023.2017. PMID: 29717933. [CrossRef] [PubMed] [Google Scholar]
- Liu L, Locascio LM, Doré S (2019), Critical role of Nrf2 in experimental ischemic stroke. Front Pharmacol 10, 153. https://doi.org/10.3389/fphar.2019.00153. PMID: 30890934. [CrossRef] [PubMed] [Google Scholar]
- Gureev AP, Popov VN (2019), Nrf2/ARE pathway as a therapeutic target for the treatment of parkinson diseases. Neurochem Res 44(10), 2273–2279. https://doi.org/10.1007/s11064-018-02711-2. PMID: 30617864. [CrossRef] [PubMed] [Google Scholar]
- Brandes MS, Gray NE (2020), NRF2 as a therapeutic target in neurodegenerative diseases. ASN Neuro 12, 1759091419899782. https://doi.org/10.1177/1759091419899782. PMID: 31964153. [CrossRef] [Google Scholar]
- Dinkova-Kostova AT, Abramov AY (2015), The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med 88(Pt B), 179–188. https://doi.org/10.1016/j.freeradbiomed.2015.04.036. PMID: 25975984. [CrossRef] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Fahey JW, Kostov RV, Kensler TW (2017), KEAP1 and done? Targeting the NRF2 pathway with sulforaphane. Trends Food Sci Technol 69(Pt B), 257–269. https://doi.org/10.1016/j.tifs.2017.02.002. PMID: 29242678. [CrossRef] [PubMed] [Google Scholar]
- Kang TC (2020), Nuclear factor-erythroid 2-related factor 2 (Nrf2) and mitochondrial dynamics/mitophagy in neurological diseases. Antioxidants (Basel) 9(7), 617. https://doi.org/10.3390/antiox9070617. PMID: 32679689. [CrossRef] [Google Scholar]
- Jeucken K, Van Hamburg JP, Tas SW (2019), Activated memory T cells produce ligands that cause Nf-Kb-dependent inflammatory activation of the endothelium: identification of novel therapeutic targets. Ann Rheum Dis 78, 285–285. https://doi.org/10.1136/annrheumdis-2019-eular.2979. [Google Scholar]
- Aoki T, Fukuda M, Nishimura M, Nozaki K, Narumiya S (2014), NF-kB as a critical factor for intracranial aneurysm formation and as a therapeutic target for treatment. Acta Neuropathol Commun. 31(2), 34. https://doi.org/10.1186/2051-5960-2-34. [CrossRef] [Google Scholar]
- Montano-Almendras C, Essaghir A, Schoemans H, Noël L, Velghe A, Varis I, Latinne D, Knoops L, Demoulin JB (2012), Phosphatidylinositol-3 kinase and NF-kB as potential therapeutic targets in myeloid malignancies associated with hypereosinophilia and PDGF receptor rearrangements. Haematologica 97(s1), 376. [Google Scholar]
- Ruike T, Striker GE, Zheng F, Leong DJ, Grosjean F, Majeska RJ, Iatridis JC, Vlassara H, Hardin JA, Cobelli NJ, Sun HB (2012), Nf-Kb: a potential mediator of adamts-5 activation and therapeutic target for cartilage breakdown in high age diet-induced osteoarthritis. Osteoarthr Cartil 20, S116–S117. https://doi.org/10.1016/j.joca.2012.02.141. [CrossRef] [Google Scholar]
- Ina K, Knsugami K, Nishiwaki T, Furuta R, Watanabe O, Suzuki T, Ando T, Goto H (2004), Suppression of endothelial Nf-kB activation may be a candidate therapeutic target in patients with ulcerative colitis. Gastroenterology 126(Wl108), A 570. [Google Scholar]
- Lynch DR, Farmer J, Hauser L, Blair IA, Wang QQ, Mesaros C, Snyder N, Boesch S, Chin M, Delatycki MB, Giunti P, Goldsberry A, Hoyle C, McBride MG, Nachbauer W, O’Grady M, Perlman S, Subramony SH, Wilmot GR, Zesiewicz T, Meyer C (2019), Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann Clin Transl Neurol 6(1), 15–26. https://doi.org/10.1002/acn3.660. PMID: 30656180. [CrossRef] [PubMed] [Google Scholar]
- Dang W (2014), The controversial world of sirtuins. Drug Discov Today Technol 12, e9–e17. https://doi.org/10.1016/j.ddtec.2012.08.003. PMID: 25027380. [CrossRef] [PubMed] [Google Scholar]
- Guarente L (1999), Diverse and dynamic functions of the sir silencing complex. Nat Genet 23(3), 281–285. https://doi.org/10.1038/15458. PMID: 10545947. [CrossRef] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L (1999), The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13(19), 2570–2580. https://doi.org/10.1101/gad.13.19.2570. PMID: 10521401. [CrossRef] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L (2001), Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410(6825), 227–230. https://doi.org/10.1038/35065638. PMID: 11242085. [CrossRef] [PubMed] [Google Scholar]
- Rogina B, Helfand SL (2004), Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A 101(45), 15998–16003. https://doi.org/10.1073/pnas.0404184101. PMID: 15520384. [CrossRef] [PubMed] [Google Scholar]
- Bonkowski MS, Sinclair DA (2016), Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17(11), 679–690. https://doi.org/10.1038/nrm.2016.93. PMID: 27552971. [CrossRef] [PubMed] [Google Scholar]
- Dai H, Sinclair DA, Ellis JL, Steegborn C (2018), Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol Ther 188, 140–154. https://doi.org/10.1016/j.pharmthera.2018.03.004. PMID: 29577959. [CrossRef] [PubMed] [Google Scholar]
- Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ (2012), Age- associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7(7), e42357. https://doi.org/10.1371/journal.pone.0042357. PMID: 22848760. [CrossRef] [PubMed] [Google Scholar]
- Massudi H, Grant R, Guillemin GJ, Braidy N (2012), NAD+ metabolism and oxidative stress: the golden nucleotide on a crown of thorns. Redox Rep 17(1), 28–46. https://doi.org/10.1179/1351000212Y.0000000001. PMID: 22340513. [CrossRef] [PubMed] [Google Scholar]
- Wang CH, Wei YH (2020), Roles of mitochondrial sirtuins in mitochondrial function, redox homeostasis, insulin resistance and type 2 diabetes. Int J Mol Sci 21(15), 5266. https://doi.org/10.3390/ijms21155266. PMID: 32722262. [CrossRef] [Google Scholar]
- Krengel U, Tornroth-Horsefield S (2015), Biochemistry. Coping with oxidative stress. Science 347(6218), 125–126. https://doi.org/10.1126/science.aaa3602. PMID: 25574006. [CrossRef] [PubMed] [Google Scholar]
- Weissig V, Guzman-Villanueva D (2015), Nanocarrier-based antioxidant therapy: promise or delusion? Expert Opin Drug Deliv 12(11), 1783–1790. https://doi.org/10.1517/17425247.2015.1063611. PMID: 26119920. [CrossRef] [PubMed] [Google Scholar]
- Loschen G, Flohé F, Chance B (1971), Respiratory Chain linked H2O2 production in pigeon heart mitochondria. FEBS Lett 18(2), 261–264. https://doi.org/10.1016/0014-5793(71)80459-3. PMID: 11946135. [CrossRef] [PubMed] [Google Scholar]
- Boveris A, Oshino N, Chance B (1972), Cellular production of hydrogen-peroxide. Biochem J 128(3), 617–630. https://doi.org/10.1042/bj1280617. PMID: 4404507. [CrossRef] [PubMed] [Google Scholar]
- Chance B, Sies H, Boveris A (1979), Hydroperoxide metabolism in mammalian organs. Physiol Rev 59(3), 527–605. https://doi.org/10.1152/physrev.1979.59.3.527. PMID: 37532. [CrossRef] [PubMed] [Google Scholar]
- Linnane AW, Eastwood H (2004), Cellular redox poise modulation; the role of coenzyme Q(10), gene and metabolic regulation. Mitochondrion 4(5–6), 779–789. https://doi.org/10.1016/j.mito.2004.07.035. PMID: 16120432. [CrossRef] [PubMed] [Google Scholar]
- Linnane AW, Eastwood H (2006), Cellular redox regulation and prooxidant signaling systems a new perspective on the free radical theory of aging. Ann N Y Acad Sci 1067, 47–55. https://doi.org/10.1196/annals.1354.008. PMID: 16803970. [CrossRef] [PubMed] [Google Scholar]
- Linnane AW, Kios M, Vitetta L (2007), Healthy aging: regulation of the metabolome by cellular redox modulation and prooxidant signaling systems: the essential roles of superoxide anion and hydrogen peroxide. Biogerontology 8(5), 445–467. https://doi.org/10.1007/s10522-007-9096-4. PMID: 17415678 Review. [CrossRef] [PubMed] [Google Scholar]
- Linnane AW, Kios M, Vitetta L (2007), The essential requirement for superoxide radical and nitric oxide formation for normal physiological function and healthy aging. Mitochondrion 7(1–2), 1–5. https://doi.org/10.1016/j.mito.2006.11.009. PMID: 17317335. [CrossRef] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD (2002), Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277(47), 44784–44790. https://doi.org/10.1074/jbc.M207217200. PMID: 12237311. [CrossRef] [PubMed] [Google Scholar]
- Nohl H, Kozlov AV, Staniek K, Gille L (2001), The multiple functions of coenzyme Q. Bioorg Chem 29(1), 1–13. https://doi.org/10.1006/bioo.2000.1193. PMID: 11300690. [CrossRef] [PubMed] [Google Scholar]
- Nohl H, Gille L, Staniek K (2005), Intracellular generation of reactive oxygen species by mitochondria. Biochem Pharmacol 69(5), 719–723. https://doi.org/10.1016/j.bcp.2004.12.002. PMID: 15710349. [CrossRef] [PubMed] [Google Scholar]
- Forman HJ, Ursini F, Maiorino M (2014), An overview of mechanisms of redox signaling. J Mol Cell Cardiol 73, 2–9. https://doi.org/10.1016/j.yjmcc.2014.01.018. PMID: 24512843. [CrossRef] [PubMed] [Google Scholar]
- Forman HJ, Maiorino M, Ursini F (2010), Signaling functions of reactive oxygen species. Biochemistry 49(5), 835–842. https://doi.org/10.1021/bi9020378. PMID: 20050630. [CrossRef] [PubMed] [Google Scholar]
- Weissig V, Guzman-Villanueva D (2015), Nanocarrier-based antioxidant therapy: promise or delusion? Expert Opin Drug Deliv 12(11), 1783–1790. https://doi.org/10.1517/17425247.2015.1063611. PMID: 26119920. [CrossRef] [PubMed] [Google Scholar]
- Liberman EA, Topaly VP, Tsofina LM, Jasaitis AA, Skulachev VP (1969), Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria. Nature 222(5198), 1076–1078. https://doi.org/10.1038/2221076a0. PMID: 5787094. [CrossRef] [PubMed] [Google Scholar]
- Chen LB (1988), Mitochondrial-membrane potential in living cells. Annu Rev Cell Biol 4, 155–181. https://doi.org/10.1146/annurev.cb.04.110188.001103. PMID: 3058159. [CrossRef] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Raganos F, Staiano-Coico L, Kapuscinski J, Melamed MR (1982), Interactions of rhodamine-123 with living cells studied by flow-cytometry. Cancer Res 42(3), 799–806. PMID: 7059978. [PubMed] [Google Scholar]
- Kubin RF, Fletcher AN (1982), Fluorescence quantum yields of some rhodamine dyes. J Lumin 27, 455–462. https://doi.org/10.1016/0022-2313(82)90045-X. [CrossRef] [Google Scholar]
- Weiss MJ, Wong JR, Ha CS, Bleday R, Salem RR, Steele GD Jr, Chen LB (1987), Dequalinium, a topical antimicrobial agent, displays anticarcinoma activity based on selective mitochondrial accumulation. Proc Natl Acad Sci U S A 84(15), 5444–5448. https://doi.org/10.1073/pnas.84.15.5444. PMID: 3474661. [CrossRef] [PubMed] [Google Scholar]
- Horobin RW, Trapp S, Weissig V (2007), Mitochondriotropics: a review of their mode of action, and their applications for drug and DNA delivery to mammalian mitochondria. J Control Release 121(3), 125–136. https://doi.org/10.1016/j.jconrel.2007.05.040. PMID: 17658192. [CrossRef] [PubMed] [Google Scholar]
- Cocheme HM, Kelso GF, James AJ, Ross MF, Trnka J, Mahendiran T, Asin-Cayuela J, Blaikie FH, Manas ARB, Porteous CM, Adlam VJ, Smith RAJ, Murphy MP (2007), Mitochondrial targeting of quinones: therapeutic implications. Mitochondrion 7(Suppl), S94–S102. https://doi.org/10.1016/j.mito.2007.02.007. PMID: 17449335. [CrossRef] [PubMed] [Google Scholar]
- Filipovska A, Kelso GF, Brown SE, Beer SM, Smith RAJ, Murphy MP (2005), Synthesis and characterization of a triphenylphosphonium-conjugated peroxidase mimetic. Insights into the interaction of ebselen with mitochondria. J Biol Chem 280(25), 24113–24126. https://doi.org/10.1074/jbc.M501148200. PMID: 15831495. [CrossRef] [PubMed] [Google Scholar]
- Ross MF, Kelso GF, Blaikie FH, James AM, Cochemé HM, Filipovska A, Da Ros T, Hurd TR, Smith RAJ, Murphy MP (2005), Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry (Mosc) 70(2), 222–230. https://doi.org/10.1007/s10541-005-0104-5. PMID: 15807662. [CrossRef] [PubMed] [Google Scholar]
- Smith RA, Kelso GF, James AM, Murphy MP (2004), Targeting coenzyme Q derivatives to mitochondria. Methods Enzymol 382, 45–67. https://doi.org/10.1016/S0076-6879(04)82003-2. PMID: 15047095. [CrossRef] [PubMed] [Google Scholar]
- Smith RA, Kelso GF, Blaikie FH, Porteous CM, Ledgerwood EC, Hughes G, James AM, Ross MF, Asin-Cayuela J, Cochemé HM, Filipovska A, Murphy MP (2003), Using mitochondria-targeted molecules to study mitochondrial radical production and its consequences. Biochem Soc Trans 31(Pt 6), 1295–1299. https://doi.org/10.1042/bst0311295. PMID: 14641046. [CrossRef] [PubMed] [Google Scholar]
- Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP (2001), Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem 276(7), 4588–4596. https://doi.org/10.1074/jbc.M009093200. PMID: 11092892. [CrossRef] [PubMed] [Google Scholar]
- Coulter CV, Kelso GF, Lin TK, Smith RA, Murphy MP (2000), Mitochondrially targeted antioxidants and thiol reagents. Free Radic Biol Med 28(10), 1547–1554. https://doi.org/10.1016/s0891-5849(00)00255-0. PMID: 10927180. [CrossRef] [PubMed] [Google Scholar]
- Smith RA, Porteous CM, Coulter CV, Murphy MP (1999), Selective targeting of an antioxidant to mitochondria. Eur J Biochem 263(3), 709–716. https://doi.org/10.1046/j.1432-1327.1999.00543.x. PMID: 10469134. [CrossRef] [PubMed] [Google Scholar]
- Szeto HH, Birk AV (2014), Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther 96(6), 672–683. https://doi.org/10.1038/clpt.2014.174. PMID: 25188726. [CrossRef] [PubMed] [Google Scholar]
- Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH (2004), Cell- permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279(33), 34682–34690. https://doi.org/10.1074/jbc.M402999200. PMID: 15178689. [CrossRef] [PubMed] [Google Scholar]
- Szeto HH (2014), First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171(8), 2029–2050. https://doi.org/10.1111/bph.12461. PMID: 24117165. [CrossRef] [PubMed] [Google Scholar]
- Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto HH (2013), The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 24(8), 1250–1261. https://doi.org/10.1681/ASN.2012121216. PMID: 23813215. [CrossRef] [PubMed] [Google Scholar]
- Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH (2014), Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol 171(8), 2017–2028. https://doi.org/10.1111/bph.12468. PMID: 24134698. [CrossRef] [PubMed] [Google Scholar]
- O’Rourke B (2007), Mitochondrial ion channels. Annu Rev Physiol 69, 19–49. https://doi.org/10.1146/annurev.physiol.69.031905.163804. PMID: 17059356. [CrossRef] [PubMed] [Google Scholar]
- Ponnalagu D, Singh H (2020), Insights into the role of mitochondrial ion channels in inflammatory response. Front Physiol 11, 258. https://doi.org/10.3389/fphys.2020.00258. PMID: 32327997; PMCID: PMC7160495. [CrossRef] [PubMed] [Google Scholar]
- Schein SJ, Colombini M, Finkelstein A (1976), Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J Membr Biol 30(2), 99–120. https://doi.org/10.1007/BF01869662. PMID: 1011248. [CrossRef] [PubMed] [Google Scholar]
- Rostovtseva TK, Tan W, Colombini M (2005), On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr 37(3), 129–142. https://doi.org/10.1007/s10863-005-6566-8. PMID: 16167170. [CrossRef] [PubMed] [Google Scholar]
- De Pinto V (2021), Renaissance of VDAC: new insights on a protein family at the interface between mitochondria and cytosol. Biomolecules 11(1), 107. https://doi.org/10.3390/biom11010107. PMID: 33467485. [CrossRef] [PubMed] [Google Scholar]
- Nicholls DG (2021), Mitochondrial proton leaks and uncoupling proteins. Biochim Biophys Acta Bioenerg 1862(7), 148428. https://doi.org/10.1016/j.bbabio.2021.148428. PMID: 33798544. [CrossRef] [PubMed] [Google Scholar]
- Ricquier D (2017), UCP1, the mitochondrial uncoupling protein of brown adipocyte: a personal contribution and a historical perspective. Biochimie 134, 3–8. https://doi.org/10.1016/j.biochi.2016.10.018. PMID: 27916641. [CrossRef] [PubMed] [Google Scholar]
- Nicholls DG, Bernson VS, Heaton GM (1978), The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Exp Suppl 32, 89–93. https://doi.org/10.1007/978-3-0348-5559-4_9. PMID: 348493. [Google Scholar]
- Bouillaud F, Raimbault S, Ricquier D (1988), The gene for rat uncoupling protein: complete sequence, structure of primary transcript and evolutionary relationship between exons. Biochem Biophys Res Commun 157(2), 783–792. https://doi.org/10.1016/s0006-291x(88)80318-8. PMID: 3202878. [CrossRef] [PubMed] [Google Scholar]
- Bouillaud F, Villarroya F, Hentz H, Raimbault S, Cassard AM, Ricquier D (1988), Detection of brown adipose tissue uncoupling protein mRNA in adult patients by a human genomic probe. Clin Sci (Lond) 75(1), 21–27. https://doi.org/10.1042/cs0750021. PMID: 3165741. [CrossRef] [PubMed] [Google Scholar]
- Ricquier D, Bouillaud F (2000), Mitochondrial uncoupling proteins: from mitochondria to the regulation of energy balance. J Physiol 529(Pt 1), 3–10. https://doi.org/10.1111/j.1469-7793.2000.00003.x. PMID: 11080246. [CrossRef] [PubMed] [Google Scholar]
- Ramsden DB, Ho PWL, Ho JWM, Liu HF, So DHF, Tse HM, Chan KH, Ho SL (2012), Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav 2(4), 468–478. https://doi.org/10.1002/brb3.55. PMID: 22950050. [CrossRef] [PubMed] [Google Scholar]
- Ho JW, Ho PWL, Liu HF, So DHF, Chan KH, Tse ZHM, Kung MHW, Ramsden DB, Ho SL (2012), UCP4 is a target effector of the NF-kappaB c-Rel prosurvival pathway against oxidative stress. Free Radic Biol Med 53(2), 383–394. https://doi.org/10.1016/j.freeradbiomed.2012.05.002. PMID: 22580300. [CrossRef] [PubMed] [Google Scholar]
- Ho PW, Ho JWM, Tse HM, So DHF, Yiu DCW, Liu HF, Chan KH, Kung MHW, Ramsden DB, Ho SL (2012), Uncoupling protein-4 (UCP4) increases ATP supply by interacting with mitochondrial complex II in neuroblastoma cells. PLoS One 7(2), e32810. https://doi.org/10.1371/journal.pone.0032810. PMID: 22427795. [CrossRef] [PubMed] [Google Scholar]
- Deluca HF, Engstrom GW (1961), Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A 47(11), 1744–1750. https://doi.org/10.1073/pnas.47.11.1744. PMID: 13885269. [CrossRef] [PubMed] [Google Scholar]
- Hoppe UC (2010), Mitochondrial calcium channels. FEBS Lett 584(10), 1975–1981. https://doi.org/10.1016/j.febslet.2010.04.017. PMID: 20388514. [CrossRef] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AJ, Mootha VK (2010), MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467(7313), 291–296. https://doi.org/10.1038/nature09358. PMID: 20693986. [CrossRef] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011), Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476(7360), 341–345. https://doi.org/10.1038/nature10234. PMID: 21685886. [CrossRef] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R (2011), A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476(7360), 336–340. https://doi.org/10.1038/nature10230. PMID: 21685888. [CrossRef] [PubMed] [Google Scholar]
- Wang Y, Han Y, She J, Nguyen NX, Mootha VK, Bai XC, Jiang Y (2020), Structural insights into the Ca(2+)-dependent gating of the human mitochondrial calcium uniporter. Elife 9, e60513. https://doi.org/10.7554/eLife.60513. PMID: 32762847. [CrossRef] [PubMed] [Google Scholar]
- Esteras N, Abramov AY (2020), Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells 9(9), 2135. https://doi.org/10.3390/cells9092135. PMID: 32967303; PMCID: PMC7564294. [CrossRef] [Google Scholar]
- Inoue I, Nagase H, Kishi K, Higuti T (1991), ATP-sensitive K+ channel in the inner membrane. Nature 352(6332), 244–247. https://doi.org/10.1038/352244a0. PMID: 1857420. [CrossRef] [PubMed] [Google Scholar]
- Siemen D, Loupatatzis C, Borecky J, Gulbins E, Lang F (1999), Ca2+-activated K channel of the BK-type in the inner mitochondrial membrane of a human glioma cell line. Biochem Biophys Res Commun 257(2), 549–554. https://doi.org/10.1006/bbrc.1999.0496. PMID: 10198249. [CrossRef] [PubMed] [Google Scholar]
- Lamping KA, Gross GJ (1985), Improved recovery of myocardial segment function following a short coronary occlusion in dogs by nicorandil, a potential new antianginal agent, and nifedipine. J Cardiovasc Pharmacol 7(1), 158–166. https://doi.org/10.1097/00005344-198501000-00026. PMID: 2580137. [CrossRef] [PubMed] [Google Scholar]
- Rotko D, Kunz WS, Szewczyk A, Kulawiak B (2020), Signaling pathways targeting mitochondrial potassium channels. Int J Biochem Cell Biol 125, 105792. https://doi.org/10.1016/j.biocel.2020.105792. PMID: 32574707. [CrossRef] [PubMed] [Google Scholar]
- Hausenloy DJ, Schulz R, Girao H, Kwak BR, De Stefani D, Rizzuto R, Bernardi P, Di Lisa F (2020), Mitochondrial ion channels as targets for cardioprotection. J Cell Mol Med 24(13), 7102–7114. https://doi.org/10.1111/jcmm.15341. PMID: 32490600. [CrossRef] [PubMed] [Google Scholar]
- Wrzosek A, Augustynek B, Żochowska M, Szewczyk A (2020), Mitochondrial potassium channels as druggable targets. Biomolecules 10(8), 1200. https://doi.org/10.3390/biom10081200. PMID: 32824877. [CrossRef] [Google Scholar]
- Wong JM, De Souza R, Kendall CWC, Emam A, Jenkins DJA (2006), Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 40(3), 235–243. https://doi.org/10.1097/00004836-200603000-00015. PMID: 16633129. [CrossRef] [PubMed] [Google Scholar]
- Weissig V, Edeas M (2021), Recent developments in mitochondrial medicine (Part 1). Fopen 4, 1–13. [Google Scholar]
- Sagan L (1967), On origin of mitosing cells. J Theor Biol 14(3), 255–274. https://doi.org/10.1016/0022-5193(67)90079-3. PMID: 11541392. [Google Scholar]
- Franco-Obregon A, Gilbert JA (2017), The microbiome-mitochondrion connection: common ancestries, common mechanisms, common goals. mSystems 2(3), e00018-17. https://doi.org/10.1128/mSystems.00018-17. PMID: 28497122. [CrossRef] [PubMed] [Google Scholar]
- Clark A, Mach N (2017), The crosstalk between the gut microbiota and mitochondria during exercise. Front Physiol 8, 319. https://doi.org/10.3389/fphys.2017.00319. PMID: 28579962. [CrossRef] [PubMed] [Google Scholar]
- Saint-Georges-Chaumet Y, Edeas M (2016), Microbiota-mitochondria inter-talk: consequence for microbiota-host interaction. Pathog Dis 74(1), ftv096. https://doi.org/10.1093/femspd/ftv096. PMID: 26500226. [CrossRef] [PubMed] [Google Scholar]
- Dache ZA, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, Arena G, Lasorsa L, Bennett A, Grange T, Messaoudi SE, Mazard T, Prevostel C, Thierry AR (2020), Blood contains circulating cell-free respiratory competent mitochondria. FASEB J 34(3), 3616–3630. https://doi.org/10.1096/fj.201901917RR. PMID: 31957088. [CrossRef] [PubMed] [Google Scholar]
- Missiroli S, Genovese I, Perrone M, Vezzani B, Vitto VAM, Giorgi C (2020), The role of mitochondria in inflammation: from cancer to neurodegenerative disorders. J Clin Med 9(3), 740. https://doi.org/10.3390/jcm9030740. [CrossRef] [Google Scholar]
- Jackson DN, Theiss AL (2020), Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes 11(3), 285–304. https://doi.org/10.1080/19490976.2019.1592421. PMID: 30913966. [CrossRef] [PubMed] [Google Scholar]
- Mottawea W, Chiang CK, Mühlbauer M, Starr AE, Butcher J, Abujamel T, Deeke SA, Brandel A, Zhou H, Shokralla S, Hajibabaei M, Singleton R, Benchimol EI, Jobin C, Mack DR, Figeys D, Stintzi A (2016), Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat Commun 7, 13419. https://doi.org/10.1038/ncomms13419. PMID: 27876802. [CrossRef] [PubMed] [Google Scholar]
- Sergi D, Naumovski N, Heilbronn LK, Abeywardena M, O’Callaghan N, Lionetti L, Luscombe-Marsh N (2019), Mitochondrial (Dys)function and insulin resistance: from pathophysiological molecular mechanisms to the impact of diet. Front Physiol 10, 532. https://doi.org/10.3389/fphys.2019.00532. PMID: 31130874. [CrossRef] [PubMed] [Google Scholar]
- Vezza T, Abad-Jiménez Z, Marti-Cabrera M, Rocha M, Víctor VM (2020), Microbiota-mitochondria inter-talk: a potential therapeutic strategy in obesity and type 2 diabetes. Antioxidants (Basel) 9(9), 848. https://doi.org/10.3390/antiox9090848. PMID: 32927712. [CrossRef] [Google Scholar]
- Anderson G (2019), Endometriosis pathoetiology and pathophysiology: roles of vitamin A, estrogen, immunity, adipocytes, gut microbiome and melatonergic pathway on mitochondria regulation. Biomol Concepts 10(1), 133–149. https://doi.org/10.1515/bmc-2019-0017. PMID: 31339848. [CrossRef] [PubMed] [Google Scholar]
- Ata B, Yildiz S, Turkgeldi E, Brocal VP, Dinleyici EC, Moya A, Urman B (2019), The endobiota study: comparison of vaginal, cervical and gut microbiota between women with stage 3/4 endometriosis and healthy controls. Sci Rep 9(1), 2204. https://doi.org/10.1038/s41598-019-39700-6. PMID: 30778155. [CrossRef] [PubMed] [Google Scholar]
- Durand PY, Nicco C, Serteyn D, Attaf D, Edeas M (2018), Microbiota quality and mitochondrial activity link with occurrence of muscle cramps in hemodialysis patients using citrate dialysate: a pilot study. Blood Purif 46(4), 301–308. https://doi.org/10.1159/000490612. PMID: 30048977. [CrossRef] [PubMed] [Google Scholar]
- Simoes-Silva L, Araujo R, Pestana M, Soares-Silva I, Sampaio-Maia B (2018), The microbiome in chronic kidney disease patients undergoing hemodialysis and peritoneal dialysis. Pharmacol Res 130, 143–151. https://doi.org/10.1016/j.phrs.2018.02.011. PMID: 29444477. [CrossRef] [PubMed] [Google Scholar]
- Paule A, Frezza D, Edeas M (2018), Microbiota and phage therapy: future challenges in medicine. Med Sci (Basel) 6(4), 86. https://doi.org/10.3390/medsci6040086. PMID: 30301167. [Google Scholar]
- Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, Arena G, Lasorsa L, Bennett A, Grange T, El Messaoudi S, Mazard T, Prevostel C, Thierry AR (2020), Blood contains circulating cell-free respiratory competent mitochondria. FASEB J 34(3), 3616–3630. https://doi.org/10.1096/fj.201901917RR. PMID: 31957088. [CrossRef] [PubMed] [Google Scholar]
- Song X, Hu W, Yu H, Wang H, Zhao Y, Korngold R, Zhao Y (2020), Existence of circulating mitochondria in human and animal peripheral blood. Int J Mol Sci 21(6), 2122. https://doi.org/10.3390/ijms21062122. PMID: 32204530. [CrossRef] [Google Scholar]
- Stier A (2021), Human blood contains circulating cell-free mitochondria, but are they really functional? Am J Physiol Endocrinol Metab 320(5), E859–E863. https://doi.org/10.1152/ajpendo.00054.2021. PMID: 33719587. [CrossRef] [PubMed] [Google Scholar]
- Yu H, Hu W, Song X, Zhao Y (2020), Immune modulation of platelet-derived mitochondria on memory CD4(+) T cells in humans. Int J Mol Sci 21(17), 6295. https://doi.org/10.3390/ijms21176295. PMID: 32878069. [CrossRef] [Google Scholar]
- Bluestone JA, Herold K, Eisenbarth G (2010), Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 464(7293), 1293–1300. https://doi.org/10.1038/nature08933. PMID: 20432533. [CrossRef] [PubMed] [Google Scholar]
- Miliotis S, Nicolalde B, Ortega M, Yepez J, Caicedo A (2019), Forms of extracellular mitochondria and their impact in health. Mitochondrion 48, 16–30. https://doi.org/10.1016/j.mito.2019.02.002. PMID: 30771504. [CrossRef] [PubMed] [Google Scholar]
- Lindqvist D, Wolkowitz OM, Picard M, Ohlsson L, Bersani FS, Fernström J, Westrin A, Hough CM, Lin J, Reus VI, Epel ES, Mellon SH (2018), Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder. Neuropsychopharmacology 43(7), 1557–1564. https://doi.org/10.1038/s41386-017-0001-9. PMID: 29453441. [CrossRef] [PubMed] [Google Scholar]
- Storci G, Bonifazi F, Garagnani P, Olivieri F, Bonafè M (2021), The role of extracellular DNA in COVID-19: clues from inflamm-aging. Ageing Res Rev 66, 101234. https://doi.org/10.1016/j.arr.2020.101234. PMID: 33321254. [CrossRef] [PubMed] [Google Scholar]
- Tiku V, Tan MW, Dikic I (2020), Mitochondrial functions in infection and immunity: (Trends in Cell Biology 30, 263-275, 2020). Trends Cell Biol 30(9), 748. https://doi.org/10.1016/j.tcb.2020.07.001. PMID: 32703641 Free PMC article. No abstract available. [CrossRef] [PubMed] [Google Scholar]
- Cao Y, Li L, Feng Z, Wan S, Huang P, Sun X, Wen F, Huang X, Ning G, Wang W (2020), Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV- 2) receptor ACE2 in different populations. Cell Discov 6, 11. https://doi.org/10.1038/s41421-020- 0147-1. PMID: 32133153. [CrossRef] [PubMed] [Google Scholar]
- Singh KK, Chaubey G, Chen JY, Suravajhala P (2020), Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am J Physiol Cell Physiol 319(2), C258–C267. https://doi.org/10.1152/ajpcell.00224.2020. PMID: 32510973. [CrossRef] [PubMed] [Google Scholar]
- Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S (2020), SARS-CoV- 2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2), 271–280.e8. https://doi.org/10.1016/j.cell.2020.02.052. PMID: 32142651. [CrossRef] [PubMed] [Google Scholar]
- Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y, Lawler LA, Christie JD, Meyer NJ, Mc Causland FR, Waikar SS, Waxman AB, Chung RT, Bueno R, Rosas IO, Fredenburgh LE, Baron RM, Christiani DC, Hunninghake GM, Choi AMK (2013), Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 10(12), e1001577. https://doi.org/10.1371/journal.pmed.1001577. PMID: 24391478. [CrossRef] [PubMed] [Google Scholar]
- Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I, Kritas SK (2020), Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents 34(2), 327–331. https://doi.org/10.23812/CONTI-E. PMID: 32171193. [PubMed] [Google Scholar]
- Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C (2020), Cytokine storm in COVID-19: the current evidence and treatment strategies. Front Immunol 11, 1708. https://doi.org/10.3389/fimmu.2020.01708. PMID: 32754163. [CrossRef] [PubMed] [Google Scholar]
- Shenoy S (2020), Coronavirus (Covid-19) sepsis: revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm Res 69(11), 1077–1085. https://doi.org/10.1007/s00011-020-01389-z. PMID: 32767095. [CrossRef] [PubMed] [Google Scholar]
- Fernandez-Ayala DJM, Navas P, López-Lluch G (2020), Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp Gerontol 142, 111147. https://doi.org/10.1016/j.exger.2020.111147. PMID: 33171276. [CrossRef] [PubMed] [Google Scholar]
- Ganji R, Reddy PH (2020), Impact of COVID-19 on mitochondrial-based immunity in aging and age-related diseases. Front Aging Neurosci 12, 614650. https://doi.org/10.3389/fnagi.2020.614650. PMID: 33510633. [Google Scholar]
- Tang N, Li D, Wang X, Sun Z (2020), Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 18(4), 844–847. https://doi.org/10.1111/jth.14768. PMID: 32073213. [CrossRef] [PubMed] [Google Scholar]
- Edeas M, Saleh J, Peyssonnaux C (2020), Iron: innocent bystander or vicious culprit in COVID-19 pathogenesis? Int J Infect Dis 97, 303–305. https://doi.org/10.1016/j.ijid.2020.05.110. PMID: 32497811. [CrossRef] [PubMed] [Google Scholar]
- Vignesh R, Swathirajan CR, Tun ZH, Rameshkumar MR, Solomon SS, Balakrishnan P (2020), Could perturbation of gut microbiota possibly exacerbate the severity of COVID-19 via cytokine storm? Front Immunol 11, 607734. https://doi.org/10.3389/fimmu.2020.607734. PMID: 33569053. [Google Scholar]
- Fanos V, Pintus MC, Pintus R, Marcialis MA (2020), Lung microbiota in the acute respiratory disease: from coronavirus to metabolomics. J Pediatr Neonatal Individ Med 9(1), e090139. https://doi.org/10.7363/090139. [Google Scholar]
- Brown GC, Nicholls DG, Cooper CE (Eds.) (1999), Mitochondria and cell death, Princeton University Press. ISBN: 0-691-05026-0. [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011), A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329), 221–225. https://doi.org/10.1038/nature09663. PMID: 21124315. [CrossRef] [PubMed] [Google Scholar]
- Murphy MP (2018), Newly made mitochondrial DNA drives inflammation. Nature 560(7717), 176–177. https://doi.org/10.1038/d41586-018-05764-z. PMID: 30076376. [CrossRef] [PubMed] [Google Scholar]
- Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T, Karin M (2018), New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560(7717), 198–203. https://doi.org/10.1038/s41586-018-0372-z. PMID: 30046112. [CrossRef] [PubMed] [Google Scholar]
- Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, Crow YJ, Rice GI, Duffy D, Tamby C, Nojima T, Munnich A, Schiff M, Ribeiro de Almeida C, Rehwinkel J, Dziembowski A, Szczesny RJ, Proudfoot NJ (2018), Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560(7717), 238–242. https://doi.org/10.1038/s41586-018-0363-0. PMID: 30046113. [CrossRef] [PubMed] [Google Scholar]
- Kuhn R (2004), Eugen Bleuler’s concepts of psychopathology. Hist Psychiatry 15(59 Pt 3), 361–366. https://doi.org/10.1177/0957154X04044603. PMID: 15386868. [CrossRef] [PubMed] [Google Scholar]
- Turner T (2007), Chlorpromazine: unlocking psychosis. BMJ 334(Suppl 1), s7. https://doi.org/10.1136/bmj.39034.609074.94. PMID: 17204765. [CrossRef] [PubMed] [Google Scholar]
- Ben-Shachar D (2002), Mitochondrial dysfunction in schizophrenia: a possible linkage to dopamine. J Neurochem 83(6), 1241–1251. https://doi.org/10.1046/j.1471-4159.2002.01263.x. PMID: 12472879. [CrossRef] [PubMed] [Google Scholar]
- Bergman O, Ben-Shachar D (2016), Mitochondrial oxidative phosphorylation system (OXPHOS) deficits in schizophrenia: possible interactions with cellular processes. Can J Psychiatry 61(8), 457–469. https://doi.org/10.1177/0706743716648290. PMID: 27412728. [CrossRef] [PubMed] [Google Scholar]
- Machado AK, Pan AY, Morgana da Silva T, Duong A, Andreazza AC (2016), Upstream pathways controlling mitochondrial function in major psychosis: a focus on bipolar disorder. Can J Psychiatry 61(8), 446–456. https://doi.org/10.1177/0706743716648297. PMID: 27310240. [CrossRef] [PubMed] [Google Scholar]
- Kolar D, Kleteckova L, Brozka H, Vales K (2021), Mini-review: brain energy metabolism and its role in animal models of depression, bipolar disorder, schizophrenia and autism. Neurosci Lett 760, 136003. https://doi.org/10.1016/j.neulet.2021.136003. [CrossRef] [PubMed] [Google Scholar]
- Anglin R (2016), Mitochondrial dysfunction in psychiatric illness. Can J Psychiatry 61(8), 444–445. https://doi.org/10.1177/0706743716646361. PMID: 27412727. [CrossRef] [PubMed] [Google Scholar]
- Bar-Yosef T, Hussein W, Yitzhaki O, Damri O, Givon L, Marom C, Gurman V, Levine J, Bersudsky Y, Agam G, Ben-Shachar D (2020), Mitochondrial function parameters as a tool for tailored drug treatment of an individual with psychosis: a proof of concept study. Sci Rep 10(1), 12258. https://doi.org/10.1038/s41598-020-69207-4. PMID: 32703977. [CrossRef] [PubMed] [Google Scholar]
Cite this article as: Weissig V & Edeas M 2022. Recent developments in mitochondrial medicine (part 2). 4open, 5, 5.
All Figures
 |
Figure 1 The nine hallmarks of aging. Redrawn based on Figure 1 in [17] |
| In the text | |
|
Figure 2 Schematic overview of factors controlling the activity of PGC-1α (adapted in a simplified form from [60].) QN, Quercetin; RL, Resveratrol; BF, Bezafibrate. |
| In the text | |
|
Figure 3 Scheme illustrating functions and effects of three different levels of the intracellular concentrations of reactive oxygen/nitrogen species (adapted from [158]) |
| In the text | |
|
Figure 4 Scheme illustrating the crosstalk between microbiota and mitochondria and its systemic impact on the host |
| In the text | |